![]()
![]()
![]()
by Rafael Armisen and Fernando Galatas
Hispanagar, S.A., Poligono Industrial de Villalonquejar
Calle López Bravo "A", 09080 Burgos, Spain
INTRODUCTION
According to the US Pharmacopeia, agar can be defined as a hydrophilic colloid extracted from certain seaweeds of the Rhodophyceae class. It is insoluble in cold water but soluble in boiling water. A 1.5% solution is clear and when it is cooled to 34-43°C it forms a firm gel which does not melt again below 85°C. It is a mixture of polysaccharides whose basic monomer is galactose. These polysaccharides can be sulphated in very variable degrees but to a lesser degree than in carrageenan. For this reason the ash content is below those of carrageenan, furcelleran (Danish agar) and others. A 5% maximum ash content is acceptable for agar although it is normally maintained between 2.5-4%.
Agar is the phycocolloid of most ancient origin. In Japan, agar is considered to have been discovered by Minoya Tarozaemon in 1658 and a monument is Shimizu-mura commemorates the first time it was manufactured. Originally, and even in the present times, it was made and sold as an extract in solution (hot) or in gel form (cold), to be used promptly in areas near the factories; the product was then known as tokoroten. Its industrialization as a dry and stable product started at the beginning of the 18th century and it has since been called kanten. The word "agar-agar", however, has a Malayan origin and agar is the most commonly accepted term, although in French- and Portuguese-speaking countries it is also called gelosa.
A Japanese legend is told about the first preparation of agar:
"A Japanese Emperor and his Royal Party were lost in the mountains during a snow storm and arriving at a small inn, they were ceremoniously treated by the innkeeper who offered them a seaweed jelly dish with their dinner. Maybe the innkeeper prepared too much jelly or the taste was not so palatable but some jelly was thrown away, freezing during the night and crumbling afterwards by thawing and draining, leaving a cracked substance of low density. The innkeeper took the residue and, to his surprise, found that by boiling it up with more water the jelly could be remade".
Agar production by modern techniques of industrial freezing was initiated in California by Matsuoka who registered his patents in 1921 and 1922 in the United States. The present manufacturing method by freezing is the classic one and derives from the American one that was developed in California during the years prior to World War II by H.H. Selby and C.K. Tseng (Selby, 1954; Selby and Wynne, 1973; Tseng, 1946). This work was supported by the American Government which wanted the country to be self sufficient in its strategic needs, especially in regard to bacteriological culture media.
Apart from the above American production, practically the only producer of this phycocolloid until World War II was the Japanese industry which has a very traditional industrial structure based on numerous small factories (about 400 factories operated simultaneously). These factories were family operated, producing a non-standardized quality, and had a high employment rate as production was not mechanized. For this reason, and in spite of the later installation of some factories of a medium to small size, only in recent times has Japan operated modern industrial plants.
During the second world war the shortage of available agar acted as an incentive for those countries with coastal resources of Gelidium sesquipedale, which is very similar to the Gelidium pacificum used by the Japanese industry. So in Portugal, Loureiro started the agar industry in Oporto while at the same time J. Mejias and F. Cabrero, in Spain, commenced the studies which led to the establishment of the important Iberian agar industry. Other European countries which did not have agarophyte seaweeds tried to prepare agar substitutes from other seaweed extracts (see Appendix).
SOURCES OF AGAR
Different seaweeds used as the raw material in agar production have given rise to products with differences in their behaviour, although they can all be included in the general definition of agar. For this reason, when agar is mentioned, it is customary to indicate its original raw material as this can affect its applications (Figure 1). Hence we talk about Gelidium agar, Gracilaria agar, Pterocladia agar, etc. To describe the product more accurately, it is usual to mention the origin of the seaweeds, since Gracilaria agar from Chile has different properties from Gracilaria agar from Argentina and Gelidium agar from Spain differs from Gelidium agar from Mexico.
Originally Gelidium agar constituted what we consider genuine agar, assigning the term agaroids to the products extracted from other seaweeds. Although these agaroids do not have the same properties as Gelidium agar, they can be used as substitutes under certain conditions. After World War II, the Japanese industry was forced to use increasing quantities of raw materials other than the traditional Gelidium pacificum or Gelidium amansii due to the growing demand of the international food industry.
An increase in the agar gel strength was obtained through improvements in the industrial process during the fifties, and the differences between the genuine Gelidium agar and the agaroids then available became clearer. The gel strength increased from 400 g/cm2 (the maximum for natural agar produced by the cottage industry) to 750 g/cm2 or more for the agar produced by industrial methods. The gel strength data refer to the Nikan-Sui method which replaced the primitive Kobe method used in the past. The Nikan method is more precise and easier to reproduce. A short description of the method is included in the section on "Properties".
The Japanese discovery of the strong alkaline methods for agar extraction (see section on "Manufacturing Processes") meant an increase of the Gracilaria agar gel strength with the subsequent utilization of seaweeds imported from South Africa to increase the raw material available.
Nowadays the world agar industry basically uses the following seaweeds:
(1) Different species of Gelidium harvested mainly in Spain, Portugal, Morocco, Japan, Korea, Mexico, France, USA, People's Republic of China, Chile and South Africa.(2) Gracilaria of different species harvested in Chile, Argentina, South Africa, Japan, Brazil, Peru, Indonesia, Philippines, People's Republic of China including Taiwan Province, India and Sri Lanka.
(3) Pterocladia capillace from Azores (Portugal) and Pterocladia lucida from New Zealand.
(4) Gelidiella from Egypt, Madagascar, India, etc.
Figure 2 shows Gracilaria and Gelidium
Other seaweeds are utilized as well, such as Ahnpheltia plicata from North Japan and the Sakhalin Islands as well as Acanthopheltis japonica, Ceramiun hypnaeordes and Ceranium boydenii (Levring, Hoppe and Schmid, 1969).
Figure 2 Agarophytes. Gracilaria and Gelidium
GEOGRAPHICAL DISTRIBUTION
The geographical distribution of agarophytes is very wide and is shown in Figure 3. Main areas are located indicating the most important classes and species. The size of the coloured areas relate to the extent of the gathering area, not the quantity of seaweeds gathered.
There are areas in which different kinds of agarophytes are gathered. This is the case in Chile, a country of exceptional resources of algae. In 1984, 6 126 tons of dried Gracilaria were gathered from its very long sea coast and exported to Japan, as well as another 5 500 tons that were used by the local industry. Simultaneously, in rocky areas, sandwiched between sandy areas where Gracilaria grows, 304 tons of dried Gelidium were gathered and exported to Japan. In countries such as India and Sri Lanka, Gracilaria and Gelidiella grow together in areas relatively close to each other. Generally Gelidium resources are being exploited quite heavily and so are those of good quality Gracilaria. At present the utilization of Gelidiella is being developed.
It is difficult to evaluate the present collection of agarophytes all over the world but since Japan has been, for a long time, the sole importing country of these seaweeds (basically needed to maintain production levels of the agar industry), Japanese statistics (Figure 4a) are very valuable in giving a true view of the situation. Note that in the Japanese statistics, Gelidium seaweeds are separated from other seaweeds. In 1984, Japan imported 678 tons of Gelidium seaweeds and 9 462 tons of "other agarophytes", mainly Gracilaria and Gelidiella. However it seems that Gelidiella is included with Gelidium in some cases, probably because Gelidiella seaweeds have been called Gelidium rigidum by some phycologists in spite of the fact that they are generally considered to be of a different class. As far as agar manufacturers are concerned, they are not Gelidium since the product obtained from then is completely different from the real Gelidium agar. The Gelidium lots assigned to the Philippines (3 tonnes) and to Indonesia (62 tonnes) are probably Gelidiella. Also Gelidium from Brazil is most probably Pterocladia which can be confused with Gelidium (no Gelidium is harvested in Brazil while some quantities of Pterocladia are).
INDUSTRIAL HARVESTING TECHNIQUES
Industrial harvesting techniques for agarophytes vary, depending on circumstances, but they can be classified as follows:
(1) gathering of seaweeds washed to the shore;(2) gathering seaweeds by cutting or rooting them out from their beds;
(3) cultivation.
Figure 3 Agarophyte seaweeds distribution map
Figure 4a Agarophyte seaweeds imported by Japan 1984
|
COUNTRY |
QUANTITY |
|
Gelidium Seaweeds: |
|
|
* North Korea |
112 MT |
|
* Taiwan |
4 MT |
|
* Philippines |
3 MT |
|
* Indonesia |
62 MT |
|
* Chile |
303 MT |
|
* Brazil |
20 MT |
|
* Madagascar |
74 MT |
|
* South Africa |
100 MT |
|
TOTAL |
678 MT |
|
Other Seaweeds: |
|
|
* North Korea |
47 MT |
|
* South Korea |
48 MT |
|
* Taiwan |
77 MT |
|
* Vietnam |
15 MT |
|
* Thailand |
3 MT |
|
* Philippines |
1,470 MT |
|
* Indonesia |
69 MT |
|
* Sri Lanka |
45 MT |
|
* Chile |
6,128 MT |
|
* Brazil |
607 MT |
|
* Argentina |
58 MT |
|
* South Africa |
895 MT |
|
TOTAL |
9,462 MT |
Figure 4b Agar production in different countries indicating the seaweeds used
|
COUNTRY |
YEAR |
GELIDIUM |
PTEROCHLADIA |
OTHER SEAWEEDS |
TOTAL |
|
Japan |
1984 |
568 MT |
- |
1,872 MT |
2,440 MT |
|
Spain |
1984 |
890 |
- |
- |
890 |
|
Chile |
1984 |
- |
- |
820 |
820 |
|
South Korea |
1984 |
600 |
- |
- |
600 |
|
Morocco |
1984 |
550 |
- |
- |
550 |
|
Portugal |
1984 |
260 |
60 |
- |
320 |
|
Taiwan |
1984 |
25 |
- |
250 |
275 |
|
Argentina |
1983 |
- |
- |
197 |
197 |
|
Indonesia |
1984 |
- |
- |
150 |
150 |
|
People's Republic of China |
1984 |
50 |
- |
90 |
140 |
|
Mexico |
1984 |
80 |
- |
- |
80 |
|
United States |
1984 |
70 |
- |
- |
70 |
|
France |
1984 |
65 |
- |
- |
65 |
|
Brazil |
1983 |
- |
- |
60 |
60 |
|
New Zealand |
1983 |
- |
26 |
- |
26 |
|
TOTALS |
|
3,158 |
86 |
3,439 |
6,683 |
"Other Seaweeds" are practically all Gracilaria
Agar quantities include natural agar, industrial agar and bacteriological agar.
In countries such as India and Vietnam there exists a small agar production but their data are not available.
Gathering of seaweeds washed to the shore. In some countries these seaweeds called "argazos", "arribazon" or "beach wash". These are dead seaweeds that, after completing their biological cycle, are separated by seasonal storms. They are gathered by hand or by mechanical means from the coast or by compressed air ejectors from boats that gather the seaweeds settled in cavities at depths of about 25 metres ("wells"). To avoid fermentation, the seaweed should be gathered shortly after it has separated from its holdfast.
Gathering seaweeds by cutting or rooting them out from their beds. This work is done with rakes or grabs handled from boats or by scuba divers who operate from boats using compressed air bottles or, more frequently, a compressor on the boat connected to the diver by a hose ("hookah"). Gelidium usually occurs on rocky beds, Gracilaria on sandy ones. In general it is feasible to operate with divers in depths between 3 and 20 metres. In Japan, for many years, the seaweeds have been gathered by diving girls or "amas" who operte from floats and dive using only their lungs. These techniques of cutting or rooting out are used exclusively in some countries and are similar to the ones used for carrageenanophytes such as Chondrus crispus and other Chondrus species (Irish Moss) or alginophytes such as Macrocystis or Laminaria, adapting the equipment in each case to the morphological characteristics of each seaweed class.
Cultivation. Nowadays the need for greater quantities of agarophytes has brought about the introduction of cultivation of Gracilaria crops, along the lines used for carrageenophytes. However this cultivation has had only limited success and there are some aspects to be solved before it can be generally adopted. At the present time, cultivation for industrial purposes is undertaken in the People's Republic of China, its Taiwan Province and it is now being initiated in Chile (Ren, Wang and Chen, 1984; Ren and Chen, 1986; Cheuh and Chen, 1982; Yang, 1982; Pizarro and Barrales, 1986; Santelices and Ugarte, 1986).
POST-HARVEST TREATMENT
The preservation of seaweeds, between the time of harvesting and their actual use by the agar manufacturer, is very important. To build a seaweed processing factory, which consumes seaweeds at the rate they are harvested, is not practical. Large scale agar manufacture makes it necessary to have available quantities of agarophytes stabilized in such a way that they can be carried long distances, at the least possible cost, and stored for a long time before processing.
The first step is preservation through dehydration, to avoid fermentation that first destroys the agar and then the seaweed. The second step is pressing the weed with a hydraulic press in bales of about 60 kg, to reduce the volume and return transportation and storage costs. Dehydration must be sufficient to guarantee the seaweed's preservation, otherwise an anaerobic fermentation will occur inside the bales causing high temperatures and even carbonization of the seaweeds during storage in warehouses. In general, the moisture content is best reduced to about 20% by natural or artificial drying. Obviously it is necessary to avoid wetting during transportation and/or storage.
In the case of Gracilaria the problem is more difficult to solve. The enzymatic hydrolysis of its agar occurs spontaneously even at relatively low moisture contents, but at variable rates depending on the Gracilaria species and its origin. Gracilaria harvested in India, Sri Lanka, Venezuela, Brazil, and generally in warm waters, has an agar (agarose) less resistant to enzymatic hydrolysis than the Chilean Gracilaria which is the most stable. Nevertheless, the stability of agar contained in Gracilaria is less than that of Gelidium; Gelidium agar can be preserved in seaweeds indefinitely provided they have been well treated.
Hydrolysis of agar contained in Gracilaria can be due to endogenous enzymes or to the growth of Bacillus cereus. Pterocladia capillacea from the Azores behaves like Gelidium but the extent of hydrolysis of seaweeds such as Gelidiella, Ahnpheltia, and others has not been described.
SEAWEED PRICES
The seaweeds imported by Japan in 1984 are shown in Figure 4a. Under the heading of "Gelidium", 678 tonnes were imported at a value of Yen 253 741 000 CIF (Yen 374 250 per tonne); the exchange rate at that time was US$ 1 = Yen 246.17 so an average CIF price was US$ 1 520 per tonne. Freight must be considered in this price, especially the high cost of the freight between countries far from Japan such as Chile (303 tonnes), Brazil (20 tonnes), Madagascar (74 tonnes) or South Africa (100 tonnes). These countries account for 73% of the seaweed imported as Gelidium. The seaweeds imported by Japan are high quality and exceptionally clean, otherwise it would not be possible to afford the freight costs. This applies to Gelidium, Gracilaria or any other agarophytes. These specifications, normally not so exact when the manufacturer is located near the harvesting place, greatly increase the harvesting and preparation costs.
The economic data for "Other Seaweeds" is more difficult to interpret because although these seaweeds are referred to as Gracilaria, they may include other agarophytes like Gelidiella, Pterocladia, Ahnpheltia, etc., with different agar contents and therefore different properties. Under the heading of "Other Seaweeds", 9 462 tonnes were imported (compared with Gelidium at 678 tonnes) with a value of Yen 2 882 525 000 (Yen 304 642 per tonne) or US$ 1 237 per tonne (CIF). The freight costs in this price must also be considered since more than 80% of the "Other Seaweeds" are imported from countries such as Chile (6 128 tonnes), Brazil (607 tonnes), Argentina (58 tonnes), and South Africa (895 tonnes).
Gracilaria is the major component of "Other Seaweeds". However two types of Gracilaria are imported and they are not distinguished from each other in the import statistics. One type is clean, dried seaweed. The other type is Gracilaria which has been subjected to a strong alkaline treatment in the exporting country; this causes alkaline hydrolysis of sulphate groups, increasing the gel strength of the agar which is eventually extracted, although the yield is reduced. This treatment is expensive and for this reason the product obtained ("Colagar") brings a higher price than the untreated dried seaweed. All of this must be borne in mind when considering the average price of the "Other Seaweeds" which is calculted from the Japanese import statistics.
EVALUATION OF AGAROPHYTES
The existing literature on the evaluation of seaweeds as industrial sources of agar is confusing because in general the contributions have come from well intentioned scientists who often are unfamiliar with specification requirements, the different grades of commercial agar and the analytical methods used.
Firstly, the evaluations have been made from seaweeds which are perfectly dry and clean, like herbal samples, and therefore their data does not have any similarity to that obtained by the manufacturers who process hundreds or thousands of tonnes of commercial seaweeds arriving in very different stages of preservation, often mixed with significant quantities of impurities such as stones, shells, sand, other seaweeds, epiphytes, as well as other products added during gathering, drying, and packing (such as land weeds, leaves, wood, plastics, etc.). The quantities of salts that remain in the seaweeds after drying is another variable.
Secondly, the evaluation is frequently made without taking into account the characteristics of the agar obtained, or by comparing it only in some parameters (for instance with a bacteriological agar sample). After making some tests without knowing the real specifications for the products, and in many cases without knowledge of the usual analytical methods, it is declared to be similar. All of this is equivalent to determining the density of a rock and, seeing that it is 3.52, deducing that the rock is diamond.
Thirdly, an agarophyte evaluation is a much longer and complicated process than the one usually carried out and published in scientific articles. In our opinion it must be initiated with a series of tests or experiments on a laboratory scale. In principle we would make several extractions, combining some preliminary treatments with different extraction conditions. The previous experience of the person who is going to chose the experiments is very important if good results are to be obtained. Under these conditions it is usual to operate with glass equipment and with quantities of about 50 g of seaweed for each test. For normal work it is necessary to have between 400 and 500 g of dried seaweed.
Next, provided promising results have been obtained and a simplified quality control test has been performed, a pilot plant run should be the next step. An evaluation performed in a laboratory can be sufficient for a scientific publication but in industry, before working in a factory, we operate a pilot plant trial with quantities between 750 g and 1 kg of dried seaweeds in conditions as similar as possible to those of the industrial process. To carry out this trial it is convenient to have about 5 kg of dried seaweed available. Using the agar obtained in the pilot plant, complete analyses are made, as well as an evaluation of the product in practical applications. Knowledge of industrial manufacturing processes for agar is needed for this evaluation to be useful, as well as experience of actual specifications required by the different markets and by the practical applications of the product.
It is essential that agarophytes be correctly evaluated before starting operations in those countries that at present are studying their algal resources. For this, as soon as the quantities of agarophytes from a part of the coast have been estimated, even approximately, the quantity and quality of the agar in the seaweed should be evaluated in terms of its practical use. For this purpose it is important to have the cooperation of experienced agar manufacturers.
A very important point to be considered is the way representative samples are taken from large areas of agarophytes. Sampling is not as easy as it may seem. To have representative samples it is necessary to follow the classical sampling procedures and take some additional special precautions. The sample must be immediately packed in strong, waterproof, well fastened bags; too often samples are received in broken packages containing extremely dried seaweeds and in many cases with significant quantities of sand outside the bag and spread, through the package.
As soon as the sample is received in the control laboratory, the impermeability of the plastic bag is verified and registered in the protocol. Next, on aliquots taken in such a way that their homogeneous composition is guaranteed, the following determinations must be made.
1. Moisture. Use a drying oven at 65°C.
2. Pure seaweed determination.
Seaweeds must be soaked in fresh water for at least two hours, with stirring, to eliminate soil and sand which are decanted, filtered, dried and weighed separately. Once the seaweeds are fully swelled the agarophytes must be manually separated from all the other materials such as rocks, shells, calcareous inclusions, other seaweeds, epiphytes, various vegetable remains, wood, plastic, etc. All these materials must be dried and weighed. Then the agarophytes are washed with water until clear (some samples, particularly Gracilaria, may contain clay). When cleaned they must be dried (in an oven at 65°C) and weighed; the percentage of the sample which is "pure seaweed" is calculated.
3. Extraction - of an aliquot part of the sample
It is impossible to assign a general extraction method valid for any agarophyte. For many years we have been industrially evaluating a large number of agarophyte batches. They have come from the five continents and include Gelidium, Gelidiella, Pterocladia and Gracilaria species. We do know that it is impossible to give a valid extraction method for any agarophyte to evaluate its yield, obtaining at the same time a standard quality which allows evaluation to be useful from the point of view of the industry.
Nevertheless, we shall try to give here certain procedures that are only evaluation methods and should not be confused with industrial processes.
It is advisable to use 50 g or more in case the agarophytes have a lower percentage of "pure seaweed" than usual. Extraction conditions (pH, temperature, pressure, time, etc.) as well as the seaweed: water ratio must be adapted in each case so as to try to obtain an extract with approximately 1% agar.
A traditional Japanese method for Gelidium is the following. The seaweed (40 g) is washed three times. It is then placed in a beaker with water (40 ml, or more if necessary, to cover the seaweed which can be flattened). Adjust to pH4 using acetic acid. After 10 minutes the temperature is increased and maintained close to the boiling point for three minutes. Water is added to bring the total volume to 800 ml; with this dilution the pH increases to approximately 6 unless it is adjusted with acetic acid or a dilute solution of caustic soda. The extraction is carried out at a temperature just below boiling point for 3-4 hours, checking the seaweed texture to determine the end of the extraction. The liquid is then filtered through a cloth and the residue is squeezed. As soon as the extract gels, it is subjected to freezing, or syneresis, and afterwards is dried and weighed.
In general Gelidium, Pterocladia or Gelidiella seaweeds can also be evaluated as follows. The seaweed is mixed with a solution of sodium carbonate (0.5%, 30 ml solution for each gram of seaweed) and held at approximately 90°C for 30 minutes, allowing the alkali to diffuse into the seaweed. The seaweed is washed with running water for 10 minutes. It is then extracted with water, 30 ml for each gram of seaweed, adjusting the pH with tartaric acid between 4.8-8.0 depending on the type of seaweed and on the extracting conditions. If extraction is done at boiling point (without pressure) it is usual to work between pH 4.8-6.0 and if it is done under pressure it will depend on the pressure used but at 127°C (1.5 kg.cm-2) it is usual to operate between pH 6-8. The solution is filtered and the product is finished, either by freezing or syneresis, and dried.
In the case of certain Gracilaria species, it is necessary to make what is called a sulfate alkaline hydrolysis, working in stronger alkaline conditions to change the L-galactose 6-sulfate into 3,6-anhydro-L-galactose. For this purpose the diffusion is made with sodium hydroxide solution (at least 0.1M) for at least one hour at a temperature between 80-97°C, but with care not to extract the agar. The extraction which follows is carried out with stirring at practically neutral pH, without pressure (95-100°C), for a very variable time depending on the type of the Gracilaria used, but it can take several hours.
Then analytical control test will be needed to verify that the agar obtained meets the physico-chemical specifications that will be explained later.
CHEMICAL STRUCTURE
Early studies of agar showed that it contained galactose, 3,6-anhydro-galactose (Hands and Peats, 1938; Percival, Somerville and Forbes, 1938) and inorganic sulfate bonded to the carbohydrate (Samec and Isajevic, 1922).
Structural studies have been based on the fractionation of agar by several methods, followed by chemical and enzymatic hydrolysis. The enzymatic hydrolysis studies of W. Yaphe have been of great importance. Subsequently the spectrochemical studies using infrared spectroscopy and nuclear magnetic resonance spectroscopy, particularly 13C n.m.r., have explained many important points in the structure of these intricate polysaccharides.
Infrared spectroscopy is the most accessible method for many laboratories. Figure 8a shows different absorption bands that have been characterized for the agar spectrum. The typical bands of a carrageenan spectrum are also shown (Figure 8b) because many of its important uses are similar to those of agar and the spectra are useful for distinguishing the two. The bands at 1 540 and at 1 640 cm-1 are especially noteworthy. They come from the proteins existing in agar and about which only a few comments have been made before. The peak at 890 cm-1 has not been identified up to the present time.
N.M.R. is of great importance when studying these structures. However the technique is difficult and it requires 13C n.m.r. equipment which only a few laboratories can afford. For this kind of work it is best to consult W. Yaphe's papers, published from 1977 - for example, Bhattacharjee, Hamer and Yaphe, (1979); Yaphe (1984); Lahaye, Rochas and Yaphe (1986).
Agar is now considered to consist of two fractions, agarose and agaropectin. These were first separated by Araki (1937) and the results were published in Japanese so they were not readily available to some research workers. For example Jones and Peats (1942) assigned a single structure to agar defining it as a long D-galactose chain residue, joined by 1,3-glycosidic links; in the proposed structure, this chain was ended by a residue of L-galactose joined to the chain at C-4 and with C-6 semi-esterified by sulfuric acid. This false structure is still mentioned in some books on natural polymers and even in recently published encyclopedias.
AGAROSE
Interest in agarose was lost until Hjerten, working under Tiselius at the University of Uppsala, began to look for an electrically neutral polysaccharide suitable for electrophoresis and chromatography. He published an improved method of separation based on the use of quaternary ammonium salts (Hjerten, 1962). A technique for agarose preparation using polyethylene glycol was reported by Russell, Mead and Polson (1964) and later this was patented with Polson (1965) named as the inventor. Both methods gave agarose of sufficient purity to allow the study of its structure.
Figure 5 shows the type, and approximate relative quantities, of the residues that can be separated from the total hydrolysis of agarose.
Figure 6 shows agarose to be a neutral, long-chain molecule formed by b -D-galactopyranose residues connected through C-1 and C-3 with 3,6-anhydro-L-galactose residues connected through C-2 and C-4. Both residues are repeated alternately. The links between the monomers have different resistance to chemical and enzymatic hydrolysis. 1,3-a links are more easily hydrolysed by enzymes (Pseudomonas atlantica) and neoagarobiose results. 1,4-b links are more easily hydrolysed by acid catalysts and yield agarobiose units. Nevertheless 1,4-b links make the polysaccharide chain particularly compact and resistant to breakage, as is found in the peptidoglycan of bacteria. The molecular weight assigned to non-degraded agarose is approximately 120 000. This weight has been determined by sedimentation measurements and it represents 400 agarbiose (or 800 hexose) units linked together.
Figure 5 Agarose hydrolysis products
Figure 7 Agaropectins hydrolysis products
Figure 8a Infrared spectrocopy on agar films
|
WAVE NUMBERS |
STRUCTURE CAUSING ABSORPTION | |
|
730 |
Carbon-sulfur links vibration. |
(Cross, 1964). |
|
750 |
Carbon-sulfur links vibration. |
(Torres-Pombo, 1972). |
|
820 |
Ester-sulfate in C-6 link vibration. (Stancioff and Stanley, 1969). | |
|
850 |
C-O-S in C-4 link vibration. (De Lestang and Lloyd, 1961; Alkahane and Izumi, 1976). | |
|
890 |
Typical Agar peak with unknown meaning. | |
|
930 |
3,6-Anhydro-galactose bridge vibration. Typical Agar peak. (Stanley, 1963) | |
|
1060 |
Ester-sulfate link vibrations. (Cross, 1964). (1) | |
|
1070 |
3,6-Anhydro-galactose bridge vibration. Typical Agar peak. (Stanley, 1963) | |
|
1180 |
Ester-sulfate link vibrations (Cross, 1964). (1) | |
|
1250 |
Ester-sulfate link vibrations, (Alkahane and Izumi, 1976). (1) | |
|
1370 |
Ester-sulfate link vibrations. (Cross, 1964). (1) | |
|
1410 |
Peak with unknown meaning. | |
|
1540 |
CO-NH peptide link vibrations. (Cristiaen, 1983). | |
|
1640 |
Amine function deformations vibrations. (Cristiaen, 1983). | |
|
1750 |
Possibly a methyl group vibration. (2) | |
|
2815 |
O-CH3 link vibrations. | |
|
2830 |
O-CH3 link vibrations. | |
NOTES:
(1) Peaks at 1060, 1180, 1250 and 1370 are produced by sulfates but the position occupied in the chain by the sulfates is not clearly seen in Agar due its low content of sulfates (< 2%).(2) Peak at 1750 not attributed up to this moment could be caused by methyl groups as Agar with 6-methyl forms a peak at 1780 cm-1.
Figure 8b Infrared spectrocopy on carrageenan films
|
WAVE NUMBER |
MOLECULAR ASSIGNMENT |
ABSORBANCY RELATIVE TO 1050 CM-1 |
||
|
KAPPA |
IOTA |
LAMBDA |
||
|
800 - 805 |
3,6-ANHYDRO-GALACTOSE 2-SULFATE. (-O-SO |
0-0'2 |
0'2-0'4 |
- |
|
810 - 820 |
GALACTOSE 6-SULFATE |
- |
- |
0'1-0'3 |
|
825 - 830 |
GALACTOSE 2-SULFATE |
- |
- |
0'2-0'4 |
|
840 - 850 |
GALACTOSE 4-SULFATE (-O-SO |
0'3-0'5 |
0'2-0'4 |
- |
|
928 - 933 |
3,6-ANHYDRO-GALACTOSE (Anhydro-galactose -C-O vibration). |
0'3-0'6 |
0'2-0'4 |
0'-0'2 |
|
1.220 - 1.260 |
ESTER SULFATE (-S=O vibration). |
0'7-1'2 |
1'2-1'6 |
1'4-2'0 |
NOTES. -
1.- A peak at 831 CM-1 wide is mentioned in the Bibliography to correspond to a 3-Sulfates mixture.-O-SOequatorial vibration on C-2 of a galactose linked in (1 ® 3) ring.
-O-SO
-O-SO2.- Carrageenans have wide and strong absorption bands in 1,000-1,100 CM-1 region which are typical in all polysacharides.
3.- Maximum absorption is given by 1,065-1.020 CM-1 for all carrageenan types (Kappa, Iota, Lambda, etc.)
This clarifies the information in Figure 6. However it should be noted that, depending on the origin of the raw material, some units of 3,6-anhydro-L-galactose are replaced by L-galactose. Also some D-galactose and L-galactose units can be methylated and it is said they can be in fact 6-0-methyl-D-galactose and 2-0-methyl-Lgalactose. This methylation, arising from the seaweed used in the process, determines the agarose gel point and therefore that of the agar it comes from. D-xylose has been found in very small quantities from hydrolysed agarose but it has not been possible to assign it a position in the structure.
Polar residues such as pyruvic and sulfuric acids are also found in small quantities. They may come from the small amounts of agaropectin lef in the agarose after its preparation but in our opinion sulfate and pyruvate groups remain linked in small quantities to the agarose structure, depending on the seaweed used in agar production. We follow the traditional definition of agaroses as those products obtained as the non-charged fraction after using a classical separation technique such as the precipitation with quaternary ammonium salts by Hjerten. On the other hand, in spite of the copious bibliography on this matter (we have seen 14 different basic methods to prepare agarose), none of the methods permits an agarose preparation free of electronegative charges. Many researchers have used two or three fractionating methods successively, in order to improve the separation and reduce the amount of electronegative groups present. In spite of all these efforts, these groups could not be eliminated. To cancel the electroendosmotic flow, which might be induced by these electronegative groups, it has been necessary to fix electropositive groups or use some other means so as to reduce the migration of cations (and their solvation water molecules) fixed to electonegative groups. Consequently we consider the agarose theoretical structure a chimerical dream to which we get closer each time by using more refined fractionation methods although perhaps, in practice, it may not exist at all in agar and the agarophyte seaweeds.
Nowadays commercial agaroses for use in biochemical separation techniques have to be chemically modified, so that their structure is different from the agarose as it is extracted from the seaweed, Phycologists should be aware that this is so, unless the manufacturer states that the original chemical structure has not been modified.
Agarose is responsible for the gelling power as we know it in agar. This is a gelation in aqueous media with a very small reactivity with cations and proteins and this differentiates agar from carrageenan.
AGAROPECTIN
Agaropectin (or better, the agaropectins) have a low gelling power in water. At the present time, a specific structure has not been assigned to the agaropectins. It is customary to say that they are formed by alternating units of D-galactose and L-galactose, and that they contain all the polar groups existing in agar.
Figure 7 shows the residues obtained by hydrolysis; among them, sulfated and pyruvate residues are evident. It has been verified that L-galactose 6-sulfate and D-galactose 4-sulfate are the major sulfate residues in agar. From small to moderate quantities of 3,6-anhydro-L-galactose have also been detected. These small quantities vary depending on the origin of the seaweed, on the harvesting season, on the treatment applied during the agar manufacturing process and on the treatment used during the agarose separation process.
The presence of 4,6-0-(1-carboxyethylidene)-D-galactose has also been verified, making the position of pyruvic acid in the structure perfectly clear. This unit is relatively important in agaropectin but in agarose it appears in much lower levels, as mentioned previously, probably because agarose has terminal units of 4,6-0-(1-carboxyethylidene)-D-galactose. The quantity of pyruvic acid in agar and agarose varies widely depending on the seaweeds used as raw material; we have verified quantities between 0.2-2.50% in agar and 0.02-1.30% in agarose. In this regard the work of Hirase (1957) is very interesting and explanatory.
These variations, that sometimes can be very important, appear even in seaweeds of the same class harvested a short distance from each other and seem to be permanent and depend on the growing locations. Over a period of several years (more than 10 in some cases) we have studied different Gelidium or Gracilaria harvesting areas in Europe, Asia and America, verifying the persistence of this phenomenon that can be caused by microclimatic differences. In our opinion the differences in cations existing in certain habitats also can be a cause. Naturally the different types and species cause differences that are very important sometimes in the agarose and agaropectin structures.
In Figure 7, D-galactose 2,6-disulfate has been included because we think we have identified it in small quantities in the agaropectins of some seaweeds grown in difficult conditions ("El Niño" phenomenon). These agaropectins had high viscosity, that was also apparent in the agar from which they came, along with a lower gelling power. In cases where this sulfated residue is found, the agaropectin and the agar have undesirable properties. Also shown in Figure 7 are D-galactose and L-galactose which appear to be modular units of agaropectin. Glucuronic acid is present only in traces (like the D-xylose found in agarose).
So while the basic structure of agaropectin consists of alternating D-galactose and L-galactose, D-galactose can be substituted by D-galactose 4-sulfate, by 4,6-0-(1-carboxyethylidene)-D-galactose in certain terminal chain positions or even possibly by D-galactose 2,6-disulfate, while part of L-galactose can be replaced by 3,6-anhydro-L-galactose. These different substitutions of the basic monosaccharide give an enormous number of possible structures.
McCandless used an immunochemical method to detect different carrageenan fractions with great sensitivity (Di Ninno and McCandless, 1978 and 1978a). A similar method might be applied to studying the different kinds of agaropectins in regard to their different seaweed origins, as well as the posible evolution of the structure of agaropectins during the life of the seaweed. To do this it is necessary to take into consideration the different fractions preceding the series of biochemical transformations produced by the algal enzymatic mechanisms which result in certain terminal fractions (one of which may be agarose). The current possibilities through monoclonal antibodies would allow an improvement of the sensitivity and selectivity of the method used by McCandless.
MANUFACTURING PROCESSES
The production of agar, bacteriological agar and agarose are considered in this section.
AGAR
Agar manufacturing processes have developed since the early freezing method was used to concentrate the extracts of agarophyte seaweeds. Whichever process is used, the following criteria should be taken into consideration. Firstly, it is necessary to obtain an extract from agarophyte seaweeds that contains the largest possible amount of the existing agar in the agarophytes. Secondly, the agar obtained should have the best possible characteristics to satisfy the standards expected for this product, especially as far as the gel strength is concerned. To achieve this it is necessary to consider the following basic points for the manufacturing process.
1. The seaweed treatment prior to extraction.
2. The control of molecular weight distribution during the extraction.
3. The removal of undesired products.
4. The need to work with large volumes of dilute extracts.
5. The economics of dehydrating the dilute extracts.
1. SEAWEED TREATMENT PRIOR TO EXTRACTION
The seaweed treatments prior to extraction are very important as they will condition to a high degree the characteristics of the agar obtained. For example Gracilaria agar was once called an agaroid because at that time Gracilaria was not preteated properly resulting in a product softer than that obtained from Gelidium. Now Gracilaria is given a strong alkaline treatment before extraction. This causes hydrolysis of sulfate groups and transforms important quantities of L-galactose 6-sulfate into 3,6-anhydro-L-galactose, thereby significantly increasing the gel strength of the agar obtained. Tagawa and Kojima (1972) say the industry uses 0.25-0.5M sodium hydroxide solution at 80-90°C for 3-5 hours. Okazaki (1971) gives more detail, showing how the treatment varies depending on the country of origin of the Gracilaria (Okazaki is a useful reference for details of all methods used in the Japanese agar industry). Yang (1982) gives references to the methods used in Taiwan Province. The treatment, also called sulfate alkaline hydrolysis, must be adapted to the class of seaweed used, to obtain as much desulfation as possible while still avoiding the yield losses that this process can cause. These losses can be very important if agar is dissolved in the alkaline solution. The way these treatments are applied is variable and constitutes a part of the manufacturing process that has to be constantly adapted, according to the changing seaweeds, as it becomes a double-edged tool that can substantially reduce the yield if it is wrongly applied.
2. CONTROL OF MOLECULAR WEIGHT DURING EXTRACTION
Agar, as it occurs in seaweed, when extracted is insoluble in cold water and also practically insoluble in hot water. It is therefore necessary to extract it using suitable pH and redox conditions so that some hydrolysis occurs, thereby increasing its solubility. During this fractionation or cracking, it is necessary to avoid the subsequent reduction, by hydrolysis, of the molecular weight of the fragments which have dissolved. As all manufacturing methods are based on agar being soluble in hot water but insoluble in cold, excessive molecular weight reduction of the agar in solution would cause reduction of yields during the process, whenever molecular weights are reached for which cold solutions are possible. On the other hand it is important to avoid molecular units, in the agarophyte residues, that are not soluble either for lack of the necessary solution time or because of an excessive molecular weight that curtails solution under the conditions of extraction.
Figure 10 attempts to clarify a complex process in a simplified way since what we are putting into solution is not only agarose, with a quite uniform chemical structure, but also a mixture of agaropectins carrying electronegative charges, with a minimum solubility temperature that is above the one for agarose. We can see in the figure that all those molecules with molecular weights below PM1 will be easily extracted from the seaweed but will be lost due to their cold water solubility. In contrast, those molecules that remain in the seaweed with molecular weight above PM2 will not be extracted and will remain with the cellulose residues after extraction. The agar manufacturer has to establish working methods that enable the preparation of a molecular weight distribution curve that avoids both losses as much as possible. An ideal result would be that shown by the middle graph of the three shown in Figure 10. It is very difficult to modify the PM1 value but it is possible to increase the PM2 by raising the water temperature in the extraction; this is done by working under pressure whenever the seaweeds permit it. Naturally the differing stabilities of agars to hydrolysis poses limits to such temperature increases.
The industrial objective aims toward narrowing the type of Gaussian curve shown in Figure 10. This reduces losses and increases the molecular weight to the corresponding maximum in the chart which is accompanied by an increase in the agar gel strength. Such considerations will be correct whenever a constant agarose-agaropectin ratio is maintained.
3. REMOVAL OF UNDESIRED PRODUCTS
During the extraction process, a myriad of undesired products will be obtained as well as agar. Such products are soluble salts, seaweed pigments, cellulose, hemicellulose and many extracts coming from impurities and foreign materials contained in the weed, since commercial seaweeds differ greatly from those with which scientists work. Therefore in order to obtain the purest possible extracts in industry, seaweeds are selected and washed carefully and subjected to previous corrective treatments in which generally an alkaline solution eliminates a large quantity of foreign substances, particularly red pigments (phycoestrine), changing the weed to a green colour. This alkaline treatment is with sodium carbonate; it is milder than the alkaline treatment with sodium hydroxide which is used to improve the gel strength of Gracilaria agar.
A careful filtration will purify the extract but this is quite a difficult operation which requires a high temperature (85-100°C) because of the extract's viscosity and high gelling power. Also cellulose and seaweed "floridean starch" residues, and even clay particles, make the filtration very difficult. Pressure filters are commonly used. Filter presses are the most useful ones, although modern factories use filters specially designed for this purpose.
Differences in the raw material greatly influence the operating methods and this makes further generalizations impossible.
Figure 10 Distribution of molecular weights in agar extracts
4. LARGE VOLUMES OF DILUTE EXTRACTS
Due to the high seaweed cost, high yields of agar are essential. However the extract concentrations range from 0,8% to 1.5% as a maximum; it is difficult to work with a more concentrated extract, for filtration as well as in the rest of the process. So the more agar that is extracted, the more water must be added to keep the concentration in the above range. This means that it is necessary to work with large volumes of extracts.
5. THE ECONOMICS OF DEHYDRATING THE DILUTE EXTRACTS
An important aspect to consider is the economics for dehydrating the large volumes of dilute extracts discussed in (4), This is a characteristic problem for this industry and its solution lies in methods based on the insolubility of agar when the extracts are cooled. Sometimes, because of lack of experience with the industry, projects are encountered in which evaporation or precipitation are recommended as the means of removing the large quantities of water from the extracts. We would first like to show why these methods are not feasible and afterwards discuss the methods actually used by the industry.
A. EVAPORATION
Starting from a 1% extract, 99 litres of water have to be eliminated for each kilogram of agar and since the latent heat enthalpy for water at 100°C is 539 kcal/kg, we need 53 361 kcal/kg (539 x 99 = 53 361). In our calculations we shall compare the heat requirements at a theoretical yield (impossible to obtain and far from the obtainable one) and consider only the heat for change of state; any heat requirement derived from specific heat will not be considered because of its small relative importance.
Working in an evaporator with liquids above a 2% concentration is impossible, problems are also posed by the gelling temperature of the extract and its large non-newtonian viscosity at temperatures close to gelling. All this prevents the thermal savings that could be gained by the use of multiple-effect vacuum evaporators.
B. PRECIPITATION
A working method similar to that used for carrageenan, using alcohol precipitation, could be considered. An economical process using this technique has not been achieved so far but the process is feasible chemically. Agar precipitation in alcohol media is more difficult than for carrageenan because the precipitate is more flocculant (has low cohesion) and is difficult to recover quantitatively. A high heat consumption is required because we have to add the heat needed to evaporate the alcohol, to the 53 361 kcal needed to evaporate the water in the mixture. In addition, the alcohol used for precipitation has to be recovered by distillation for reuse.
If we make our calculations using isopropanol, which is used for producing carrageenan for economic reasons, and consider that we start with an azeotropic mixture, previously recovered, of 87% by weight we are forced to work at least 3 litres of azeotropic isopropanol for each litre of extract to be precipitated. Assuming that such a mixture has a density of 0.8234 kg/L, then for each kilo of agar it would be necessary to evaporate:
99 kg of water extract;
22.5 kg of azeotropic isopropanol.
This second item is composed of 213 kg of isopropanol and 41.5 kg of water. The latent heat enthalpy for isopropanol is 175.8 kcal/kg. Therefore the theoretical heat energy consumption would be:
water: 539 x (99 + 41.5) = 75 729 kcal;
isopropanol: 175.8 x 213 = 37 445 kcal.
This means an energy consumption of 113 174 kcal/kg of agar which is double the heat energy need to evaporate the water contained in the extract, and all of this without taking into consideration the need to concentrate the used alcohol back to the azeotrope plus the recovery of isopropanol vapors that entail a considerable amount of energy. From this it can be seen that the precipitation/dehydration process analogous to that used for carrageenan has a high energy consumption when applied to agar.
For agar, concentration methods are based on its insolubility when cooled and are used in all factories according to two basic principles: freezing or syneresis under pressure.
C. FREEZING
This consists of freezing and thawing the extract, previously gelled, and profiting from the insolubility of agar in the cold to eliminate the greatest part of the water contained in the extract. Freezing should be slow, to allow both the growth of ice crystals and the separation of agar in the highest possible concentration; this is usually followed by draining with a water-extracting centrifuge. Only slow freezing permits large ice crystals to be formed, surrounded by fine sheets of agar. Efforts to speed up freezing produce spongy masses, with high water content and less agar concentration, that dialyse poorly and produce an impure agar, because the impurities which are soluble in cold water do not move so well from the gel to the water.
As far as the economics for this process are concerned, we should consider that if we start with a 1% agar extract, we have to eliminate 99 litres of water per kg of agar; after melting and draining, this agar at best reaches a dry extract content of 15% (1 kg in 6.66 L) but is normally 11-12%. Presuming a 15% agar in the product, the cycle of freezing-defrosting eliminates (99 L - 6.66 L) 92.34 litres water per kg of agar. Furthermore the energy consumption for freezing 1 L or 1 kg of water is 79.67 kcal. To freeze the 99 litres of water contained in the 1% extract would require:
99 x 79.67 = 7 887 kcal
To remove the water remaining in the melted and drained agar requires a heat consumption of:
6.66 x 5 390 = 3 590 kcal
We can see the difference between the sum of these figures (11 477) and those for:
evaporation method = 53 361 kcal,
precipitation method = 113 174 kcal.
Naturally there is a cost difference between obtaining a difference of a kilocalorie by heating or cooling but the figures leave us in no doubt (even though we have ignored the energy consumption derived from the specific heat of the water that is eliminated) there are enormous energy differences between the working methods considered.
D. SYNERESIS
Syneresis is usually described as the process in which a gel contracts on standing and exudes a liquid. Here the term syneresis is used to describe the process where pressure is used to exude liquid from the gel. The water that soaks the colloidal net of the gel is eliminated by applying, by suitable means, a force that will favour such loss. Energy consumption is very low when working in these conditions but not everybody can benefit from it because the industrial technology is not simple. Pressure has to be applied very carefully to avoid gel losses by extruding the gel through the containing system. The advanced factories that use this process have been obliged to develop a very specific technology, not only producing extracts in the appropriate conditions for good syneresis but also equipment design that will allow the efficient treatment of large quantities of extracts.
Initially long syneresis periods were required, with cycles longer than 24 hours, that would start with a gradual and slow increase in pressure by placing, successively and at a prefixed rate, stone blocks on top of the gel containers; the agar gel was wrapped in canvas cloths and placed in a series of steel boxes fitted between the fixed and movable heads of a vertical hydraulic press. This treatment was followed by hydraulic pressing, once the product was consistent enough to withstand extrusion. Usually a modified platen press is used which is similar to a box press but the cloth bags are not enclosed on the sides during pressing and the press is usually built in horizontal form. Nowadays some agar manufacturers have designed their own modern equipment which permits this syneresis to be carried out automatically in relatively short periods of time and operating with large volumes.
Starting with a 1% agar extract, syneresis increases concentration to a maximum of 25% (1 kg agar per 3 L water). If we consider an average of 20% for the dried extract from industrial runs, the heat energy necessary to remove the rest of the water will be:
4 x 539 = 2 156 kcal/kg
which is much less than the heat energy needed to dry the agar obtained by freezing where moisture was calculated in ideal conditions, that are difficult to obtain in reality.
Compared to freezing, syneresis results in large electrical energy savings as the electrical energy needed to maintain a pressure on a quite incompressible product is much less than that necessary to freeze 99 litres of water for each kg of agar produced. The cost of electrical energy makes many freezing factories increase extract concentration but this is possible only up to 1.5% before producing a harmful effect from yield losses. Syneresis, when properly applied, will also produce a purer agar, eliminating a larger quantity of soluble matter.
6. GENERAL
Figure 11 is a flow chart showing the steps used in both of the dehydration processes used to produce agar. Treatment and reagents used in each case will be very variable depending on the species of seaweed used, its origin and even the time of the year when it was harvested. All these factors can cause drastic modifications to the treatment.
Figure 11 Agar production diagram
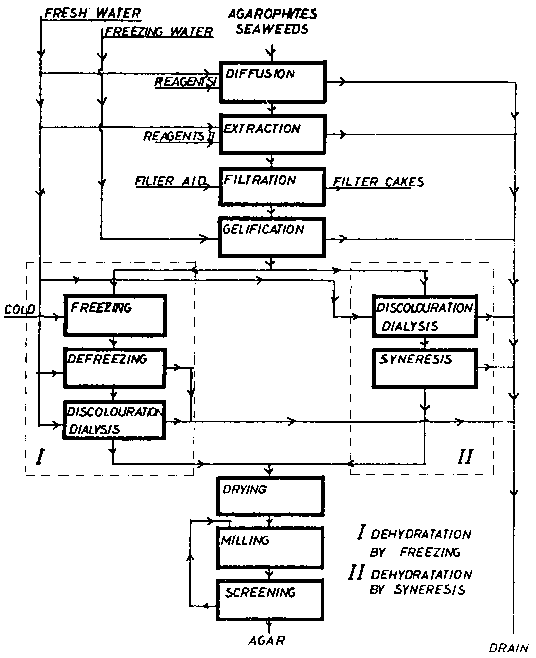
Nevertheless, we should consider some general rules. Seaweeds such as Gelidium, Pterocladia and Gelidiela can be Created by different diffusions, the most usual ones being sodium carbonate solutions, at about 80-95°C. Other reagents such as calcium or aluminium hydroxides or salts can also be used for several purposes. Other treatments with sodium hydroxide solutions of very variable concentration can be used, but the concentration will vary depending on what purposes they are for. As far as Gracilaria is concerned, 0.1 M sodium hydroxide solutions are commonly used; higher concentrations can also be used. The reagents named "Reagents I" in Figure 11 are basically the ones mentioned above. The ones shown as "Reagents II" are Chose used to adjust the extracting conditions and, in general, are organic or inorganic acids or salts with which pH and other extraction parameters are fixed.
The variables in the manufacturing process make it hard for a factory to change the seaweeds it uses as raw materials. Agar manufacturing history is full of fiascos caused by industries trying to change their seaweeds without having adequate technology to adapt to the change.
Water consumption in an agar factory varies widely depending on the seaweed used but it is always very high. Normally factories working Gracilaria seaweeds have a higher water consumption than others. Consumption also increases when an agar of better quality is required, although, in general, it can be reduced by a suitable design of the factory; however this can lead to an increase in investment and therefore to a more difficult project profitability. Factories using the freezing process have very high water consumption as cooling water is needed for the freezing equipment.
Using recycled water, after appropriate treatment, would reduce its consumption but, in general, would increase the plant operating cost. If poor quality water is going to be used, a prior treatment will be required but it is very important to know its cost before the location is decided since a mistake in this point could make the operation of the factory economically impossible.
The above-mentioned problems about water, and those originating from changing to seaweeds of a different origin, are the ones which have led many factories to bankruptcy.
A manufacturer of good quality agar must be ready to monitor his process and so be able to spot readily any variations that seaweeds cause in the yield or in the quality of the final product. For this purpose a well equipped control laboratory is required together with a pilot plant that will enable any modifications needed in the process to be studied prior to the industrial treatment of each batch of raw material. An adequate pilot plant can process from 1-10 kg of seaweeds, depending on the size and importance of the factory. In general small factories with elementary technology do not achieve international quality standards and their products have to be sold at lower prices in local markets. Bacteriological contamination particularly is usually too high and sometimes dangerous in such plants, closing them to many markets.
A food grade agar should have a moisture content of less than 18%, ash below 5%, gel strength above 750 g/cm2 (Nikan-Sui method) and a bacterial count below 10 000 bacteria per gram. Escherichia coli and Salmonella must be absent (other pathogenic bacteria may also be specified). Usually the lead content is specified as less than 5 ppm and arsenic less than 3 ppm. These specifications are for agar produced on an industrial scale. In the Orient, large quantities of "natural agar" are sold by very small producers and consumed in the form of threads ("strip") or bars ("square") that are usually produced from Gelidium and do not have to meet the above-mentioned specifications. Generally its gel strength is 450 g/cm2 by the Nikan-Sui method.
Figure 4b shows as closely as possible what we consider the present situation for the world production of agar. This table has been prepared taking into account the results obtained from an enquiry made among the most important agar manufacturers in countries such as Spain, Chile, Morocco, Portugal, Argentina, Mexico, France, New Zealand, Brazil, etc., and the available Japanese statistics. All these data along with others from Korea, People's Republic of China, its Taiwan Province and Indonesia have been updated during the XIIth International Seaweed Symposium held in Brazil, August 1986.
BACTERIOLOGICAL AGAR
The use of agar in bacteriology is one of the most important uses and requires strict physical-chemical control as well as the absence of hemolytic substances and what is more important and difficult, the absence of any bacterial inhibitors. Robert Koch started using agar in 1881 to gel culture broths when preparing solid culture media and this was the first introduction of this oriental product to Europe.
Its uses in microbiology are based on the special properties: a gelling temperature of 32-36°C, a melting temperature of 85-86°C, a lack of hydrolysis by bacterial exoenzymes and its ability to be prepared without bacterial inhibitors. The above temperatures refer to culture media gelled with agar and which contain 10-11 g agar per litre of culture media.
Bacteriological agar is prepared from Gelidium and Pterocladia because Gracilaria and Gelidiella give agars with gelling temperatures above 41°C. It is manufactured in a limited number of highly specialized factories and under rigid physical-chemical and bacteriological controls.
There are no specifications for a universal application for bacteriological agar as the different microbiological schools evaluate the parameters in various ways. There are neither international nor national specifications. There are many differences between food grade and bacteriological grade, in physico-chemical and bacteriological controls, but this information is confidential and is shared only by the bacteriological agar and culture media manufacturers.
As agar is used only as a gelling agent in solid media, it is essential to avoid interactions with the rest of the media components such as meat extract, peptones, proteins, amino acids, sugars and other carbohydrates, as well as pigments, indicators, inhibitors, mineral salts, etc., used in their formulation. It has to mix with these components without producing problems such as colour changes, precipitate formation or gel strength losses, even after autoclave sterilization. Therefore actual specifications are different depending on each user and each culture media manufacturer. In general, bacteriological agars are very transparent agars in solution as well as in gel form and they represent the purest qualities in the world market. The rest of the parameters vary as the agars are adapted to the individual requirements of the manufacturer and end user.
In much smaller quantities, and at a much higher price, another type of agar called "Purified Agar" is also available. These are bacteriological agars that could also be used in biochemistry for electrophoresis or immunodiffusion; they can be considered as agarose forerunners, being still used for economic reasons.
AGAROSE
The composition of this agar fraction has already been explained in the section dealing with the chemical structures of agar. In the literature we have found that agarose had been prepared according to at least 15 basic principles starting with the acetylation procedure of Araki (1937). A list of these methods follows even though they are interesting mainly for historical reasons.
1. Acetylation. This method is based on the different solubility in chloroform of the acetates of agarose and agaropectin.2. Selective solution. This is based on solubility differences between agarose (less soluble) and agaropectin (more soluble) in aqueous media in well established conditions.
3. Quaternary ammonium precipitation. This is classical method worked out by Hjerten (1962) and based on the insolubility of products resulting from the reaction of agaropectin with some quaternary ammonium salts.
4. Polyethylene glycol. The classical method of Polson (Russell, Mead and Polson, 1964; Polson, 1965) based on the reduced solubility of agarose in media that contain polyethylene glycol.
5. Dimethyl sulfoxide extraction. Tagawa (1966), the method is based on the different solubilities of agarose and agaropectin in this solvent.
6. Ammonium sulfate precipitation. Azhitskii and Kobozev (1967), the method is based on the precipitation of agaropectin with ammonium sulfate.
7. Ion exchange. Zabin (1969), the method is based on ion exchange in citrate or acetate forms.
8. Insoluble support absorption, Barteling (1969), the method is based on the absorption of agaropectin on a non-reactive support such as aluminium hydroxide gel.
10. Chromatography. Izumi (1970), the method is based on a chromatographic separation of agarose and agaropectin.
11. Acrinol precipitation. Fuse and Gotto (1971).
12. Electrophoresis. Hjerten (1971), the method uses electrophoresis over granulated or non-granulated agar gels or over powdered agar.
13. Rivanol precipitation. Svridov, Berdnikov and Ivanov (1971), the method depends on the precipitation of agaropectin with rivanol.
14. Chitin and chitosan precipitation. Allan et al. (1971), this method uses the absorbent chitin or chitosan to eliminate agaropectin.
15. Ethanol or 2-methoxyethanol precipitation of agarose dissolved in a urea buffer. Patil and Kale (1973).
Based largely on these methods, other publications and patents have appeared modifying or maintaining these principles for processes for the preparation of agarose. Sometimes two or even three fractionating methods have been used successively in attempts to improve the agarose quality. At this time we have records of four companies manufacturing agarose and only one of them is an agar manufacturer, very different equipment is needed for the two kinds of production. For agarose, a quality control laboratory with very sophisticated analytical equipment to analyse the finished product is essential. Continuous improvement in technology is essential to adapt to modern applications in biochemistry which have required the introduction of modifications in the chemical structure of agarose, by synthetic organic chemistry in many cases. Thus, an agarose sample obtained from a manufacturer of biochemical reagents does not correspond normally to what we can extract from agar by any of the methods previously mentioned.
Criteria for judging agarose are multiple and they can be grouped in the following way.
A. Physico-chemical properties. In this case the same basic criteria as for agar are followed: colour, transparency in solution, moisture, ash (in this case much lower due to the absence of polar groups), gel strength, gelling and melting temperatures.B. Purity critera. Reduction of electronegative groups to the minimum, the effects of such groups include an electroendosmosis increase and also an increase in the fixation of electrically charged substances, such as an increase in non-selective fixation of proteins. The increased presence of electronegative groups can also be produced by poor separation from the agaropectins. Likewise it is very important to assure the absence of residues of reagents used in the agarose production process.
C. Specifications are necessary for practical applications, such as protein electrophoresis, DNA residues, non-selective fixation of proteins. For example the absence of inhibitors that could hinder the DNA recombining fragments split by agarose techniques. Controls that will prove agarose to be acceptable for biochemical techniques are included in this group.
Generally, the first two groups appear in specifications even though in some cases the data offered causes confusion, as happens for example in electroendosmosis. Although an accepted criteria for purity is a low electroendosmosis (less electronegative groups present) there are agaroses that have a greater electroendosmosis and yet are better in some specific biochemical separations. Values given to electroendosmosis vary widely for the same agarose when analytical conditions change, such as buffer pH, ionic strength, protein standards and non-charged molecules as well as other conditions dependent on the equipment such as voltage, operating cycle, refrigeration or electrical contact strips. The growing biochemical applications of agarose imply modifications in its structure to expand its range of uses. Thus, it is not realistic to set detailed specifications for a continuously evolving product and none have been set at a national or international level. Some typical specifications for commercial agarose can be found in the Sigma Catalogue (Sigma Chemical Co. 1987) and FMC offer their analytical methods to scientists in their catalogue, "Marine Colloids 1981 Bioproducts Catalog".
PROPERTIES
The most important characteristics of agar are the following.
1. Its great gelling power in an aqueous environment allows it to form gels which are more resistant (stronger) than those of any other gel-forming agent, assuming the use of equal concentrations.2. The simple water solution has that gelling power. There is no need to add reagents to produce gelation, such as potassium (or proteins as is necessary with carrageenans), calcium (or other divalent cations as is necessary with alginates). High sugar concentrations or an acid environment (as is necessary with pectins) are not needed.
3. It can be used over a wide range of pH, from 5 to 8, and in some cases beyond these limits.
4. It withstands thermal treatments very well, even above 100°C which allows good sterilization.
5. A 1.5% aqueous solution gels between 32°C-43°C and does not melt below 85°C. This is a unique property of agar, compared to other gelling agents.
6. Agar gives gels without flavour and does not need the additions of cations with strong flavours (potassium or calcium), it can be used without problems to gel food products with soft flavours.
7. It assimilates and enhances flavours of products mixed with it and acts as a fragrance-fixer permitting their long term fixation.
8. Its gel has an excellent reversibility allowing it to be repeatedly gelled and melted without losing any of the original properties.
9. Transparent gels that are easily coloured can be obtained whose refractive index can also be easily increased by adding sugar, glucose, glycerine, etc., given them an attractive brightness.
10. The gel is very stable, not causing precipitates in the presence of certain cations as happens to alginates with calcium.
The gelling properties of agar are the origin of its multiple applications; it has the highest natural gel strength of any gelling agent. Therefore the methods used to measure its gel strength are important, as is a knowledge of the structure of the gel. Both these matters will be discussed.
MEASURING GEL STRENGTH
The following methods for measuring gel strength are basic controls in the agar market yet they are not mentioned in the Pharmacopeias, National Formulary, Codex or other similar publications of specifications and analytical methods referring to agar. This results in confusion as these methods are generally used for food grade agar, bacteriological agar and for agarose, by industry and commerce.
The Nikan-Sui method is the most common one used to measure the agar gel strengh. This method is based on measuring the load (g.cm-2) that causes a standard gel to break in 20 seconds. A hot 1.5% solution is poured into metallic boxes (6 x 30 cm base, 4.5 cm high) to the 3 cm level, leaving it to gel at 20°C. The breaking load withstood for 20 seconds is measured with an apparatus designed by the engineer Takenami and made by KIYA SEISAKUSHO LTD., 50 Komagomo, oiwake-cho, Bunku, Tokyo, Japan, (see Figure 12). The solution is made in such a way that total solution of the agar and a final concentration of 1.5% are guaranteed. Boiling and stirring must be maintained long enough to avoid the agar sticking to the bottom of the box; this is achieved by boiling under reflux or by adding hot water to maintain the initial weight and so keep the concentration constant. The load used is a cylindrical plunger with a frontal area of 1 cm2.
Another technique used in some markets is based on a Rowerbal weighing machine (Figure 13) which adds increasing loads until the gel ruptures. The gel, 1.5% agar, is prepared in a similar way to the one used for the Nikan-Sui method but in a crystallizing dish (70 mm diameter, 52 mm high) to a level of 48 mm. The value obtained by this method differ a little from the ones obtained by the Nikan method, so it is always important to state the method used for gel strength control.
The gel strength of a 1.5% solution of industrial agar lies between 600 and 1 100 g.cm-2 (Nikan-Sui method); the strength of the normal quality is 700-800 g.cm-2. This is between five and eight times higher than the gel strength of other colloids used in the food industry.
GEL FORMATION AND STRUCTURE
In agar gels, helicoidal structures have been verified by X-ray diffraction, similar to those found in carrageenan. However because agar contains 3,6-anhydro-L-galactose, the helices are left-handed whereas in kappa and iota carrageenan, which contains 3,6-anhydro-D-galactose, they are right-handed (dextrogyres). Also the helix pitch is shorter than the 26A° of carrageenan. This is explained by several authors (Arnott et al., 1974) as being due to the lower content of sulfate groups that possibly cause a tighter and more compact net. Molecular configuration changes and agarose interaction in sol-gel transitions have been well studied through ultra vacuum circular dichroism (u.c.v.d).
The gelation process from solution (colloidal sol) happens as per the scheme shown in Figure 9 which shows the different steps starting from random coil, through the left-handed dual helix formed by hydrogen bond formation chat then will be the base for the macro grid that will give the gel rigidity. The hydrogen bond formation can be obstructed and even prevented if a proton-catching agent (like urea or guanidine) is added to the sol to be gelled. Such additions prevent agar or agarose gelation and result in a solution, similar to glycerin, which when cooled does not gel. By removing the proton catcher, the hydrogen bonds will form and therefore the gel-forming ability will be restored. In the same way, considering that dry agar, be it in powder, flake, square or strip from, is really a dry gel (xerogel), its solubility in the cold is not possible as it maintains the hydrogen bonds formed during the gelation prior to its dehydration.
Figure 12 Gel strength measurement. Nikan-Sui Method
Figure 13 Gel strength measurement. Rowerbal method
A fundamental characteristic of an agar and agarose gel is what can be called "gelation hysteresis". An agar or agarose gel, when cooled, forms a gel at temperatures between 32° and 43°C depending on the seaweeds used, as that will determine the presence of a variable quantity of methyl groups. However when the well formed gel is heated, a temperature of 85°C must be reached to get the gel to melt and to become a sol. Such a big difference between gelling and melting temperatures is exceptional when compared with the rest of the phycocolloids. It is explained by a greater number of hydrogen bonds and the lack of sulfate groups, which produce a gel with helix pitches much shorter than those of carrageenans, and that, in contrast, does not show cation reactivity.
The characteristic of "viscosity hysteresis", is also remarkable. This can be demonstrated by a solution or colloidal sol prepared at boiling point and held in a thermostatic bath, for example at 80°C, and then its viscosity measured. Afterwards it is held at a lower temperature, at 50°C for example (above the gelling temperature of the sol) keeping it there for a few hours. Subsequently it is held again at 80°C and once this temperature is reached its viscosity is determined again, and it gives values higher than those initially measured. When this temperature is maintained, viscosity values decline slowly almost down to the values measured the first time. Therefore the viscosity values obtained for a solution of agar could depend on its previous history.
USES
Agar was the first phycocolloid to be used in the human food industry. In the beginning it was only used in the Far East, but the applications have been extending all over the world for more than a century. The increasing range of applications is due to the particular gelling characteristics which are not present in any other phycololloid, gum or gelatin. As a result the price for food grade agar is higher than that of other phycocolloids with gelling properties which are also permitted as food additives. In addition, these characteristics allow agar to be used successfully and even exclusively in certain scientific and industrial applications. Some earlier reviews have also discussed uses of agar (Selby and Wynne, 1973; Meer, 1980; Glicksman, 1983).
The 10 important characteristics of agar, listed at the beginning of the "Properties" section, explain technically many of the applications of agar. For use by the human food industry its safety is guaranteed by more than three hundred years of non-interrupted use by some countries and for more than a century on a world scale. In addition the FAO/WHO Codex Alimentarius permits the use of agar in the human food industry and it has also been accepted and authorized by the regulations of the more exacting countries such as United Kingdom, Federal Republic of Germany, Russia, France and Poland. The Food and Drug Administration (FDA) of the United States assigns agar as a grading of GRAS (Generally Recognised as Safe).
In the human food industry, agar is used mainly as a gelling agent and in a secondary way as a stabilizing agent and for controlling viscosity. It is used as an additive, not as a nutrient. The gelling power of agar is so high that it is used at 1% maximum concentration; for viscosity control and as a stabilizing agent the proportion used is 1/100 or less. For this reason the ingested quantities are very small and, because agar is not easily digested by the human body, its calorie contribution is negligible and thus agar can be used in special diet food. Agar digestion by the human body is imperfect, studies have shown that less than 10% of the polysaccharide is assimilated. Therefore due to the small proportions in which it is used in human food, its importance as a nutrient is very small.
Agar applications in the food industry are based on its special characteristics and the most important applications are the following.
In confectionery, to prepare jellies, marshmallows and candies or candy fillers.
In marmalade production, agar is used as a thickening and gelling agent.
Mitsumame production in Japan is very important; this is a fruit salad mixed with agar gel cubes, duly coloured, salted and flavoured with fruit flavour. The agar used for this kind of fruit salad must allow the cans to be sterilized without the cubes melting or losing their corners or edges. For this purpose certain types of Gelidium agar are used.
In bakery, agar is used to cover cakes, in icing doughnuts, and when it is applied to chocolate it allows a good adherence to the base without cracking. In general agar is utilized to prevent dehydration of these confectionery products.
Agar is also important in fruit jelly preparations. When compared with pectin, agar has the advantage of not needing high sugar concentrations to form a gel.
Its application in yoghurt is also very important especially when consumers started to require less acid products and, therefore, casein cannot contribute to the maintenance of the product consistency, as it previously did.
In the meat industry, and especially in the preparation of soft boiled sausages, its use has permitted the reduction of fat content that acted before as bonding. Today the industry is trying to limit fat content in order to reduce cholesterol.
Agar is also used on a large scale in canned products like "scatola" meat (beef blocks in gelatine) - very popular in Italy, or chicken in gelatine - very common in Canada, cow tongue in gelatine -selling well in Denmark, lamb tongue in Australia, or other different types of meat and fish aspics. In dressings and extracts it is used as a thickener and stabilizer.
In smaller quantities, agar is used to increase the viscosity of some alcoholic liquers.
The important gel-forming properties of agar have permitted the expansion of its use to applications other than the human food industry. To prepare casting moulds, an aqueous solution of high concentration (8% or more) is used with the addition of glycerin or glycols, as well as a preservative to avoid gel surface contamination by moulds; bacterial contamination is impossible because a gel with such a high concentration has very little free water left where bacteria can grow (it has a very low AW). These kinds of gels are also used in sculpture, archaeology, and in other works in which a perfect or precise reproduction is essential. For this reason it is also used in dental moulds as it is possible to make better and more precise reproductions, in spite of the fact that casting moulds prepared with agar are more expensive than those prepared with alginate paste. The purpose of adding glycerin in these uses is to avoid cast dehydration since a balance with the outside humidity can be achieved and therefore stable gels can also be obtained which do not appreciably change the gelling and melting points. Glycerin also expedites heat transfer permitting a faster gel melting in a boiling water-bath. The perfect reversibility of an agar gel permits it to be repeatedly used in this application and its low gel point makes it possible to be used as a dental mould.
Another rare application is the preparation of food to feed insects during their larval stages. The breeding of certain insects on a large scale, such as the Mediterranean fruit fly that attacks fruit trees or Pectinopora glosipeii that attacks cotton plantations, has made plague control possible through a type of biological warfare. Large numbers of insects are grown and sexually sterilized by gamma rays. When released they drastically reduce the reproduction of the overall population.
The application of agar in pharmacy as a smooth laxative is well known. Lately it has been used as an excipient in pharmaceutical preparations. In some Western countries agar is used as an antirheumatic since a prolonged treatment has permitted important improvements in patients' health. Agar has been used to stabilize cholesterol solutions.
Due to its low tendency to precipitate in alcoholic media, it is possible to prepare agar gels with alcohol concentrations that will burn when a match is applied to them.
Agar has been used in orchid nurseries for a long time. Improvement in cellular cultivation know-how has brought another important application of agar mainly in the techniques that, starting from meristems, produce perfect and virus-free clones of plants. Agar for these more refined agricultural applications must be especially prepared and strictly controlled to guarantee the absence of inhibitors chat prevent cellular cultivation, or make it difficult. This use is increasing since cellular cultivation media for vegetables is being used in industrial quantities because of the current application of these techniques in agriculture.
The 10 basic points previously mentioned are very important as far as Bacteriological Agar and Agarose applications are concerned, as well as those properties essential for the applications of both products. Agarose is produced from both Gelidium and Gracilaria and these two raw materials can give agaroses with different properties which are useful in various applications. Agarose is employed in biochemical technology for protein separation, mainly in analytical laboratories, but has started to appear in industrial applications due to the impressive growth of the genetic engineering industry that produces proteins such as interferon, interleukin and insulin which are sometimes separated with beads made of agarose. Microbiologists employ it in tissue culture as well as in other very sophisticated techniques. Renn (1984) has described some of the applications of agarose.
The uses of agarose can be grouped in the following broad categories.
1. Immunodiffusion and diffusion techniques2. Electrophoresis of particles carrying electrical charges with direct application for proteins, nucleic acids, polysaccharides that necessarily are charged in conventional electrophoresis as well as reverse electrophoresis, immunoelectrophoresis or electrofocusing.
3. Chromatographic techniques in gel chromatography, ion exchange chromatography (for which the required polar groups will be fixed in the agarose), affinity chromatography or chromatofocusing.
4. In bioengineering as a raw material for beads used in chromatographic columns for separations of proteins, as well as cross-linked beads to which active molecules can be attached which can be recovered afterwards.
5. In microbiology as an excellent base for growing very special cultures, in many cases related to oncological research.
It can be seen that agarose uses are developing as biochemistry and especially bioengineering advance. It constitutes an inert support of natural origin, modifiable by organic synthesis, with the highest known gelling power among natural colloids. Moreover it is of such a great stability that no reagents are needed to preserve it indefinitely, thus avoiding foreign interference with the products meant to be separated.
MARKETING
Agar is marketed pure, without adding mineral salts, other gums or products destined to standardize its properties as is done with carrageenans and some alginates. For that reason, it is very important to know the different techniques of gel strength measurement since the industry produces agars of different gel strengths. The use of agar combinations with other gums for several applications, to solve different rheological problems, makes it necessary to explain here its behaviour when mixed with substances to obtain certain textures. In some applications, agar by itself gives a brittle texture and to improve its elasticity, it is mixed with locust bean gum (also called carob gum) to obtain more elastic gels. In this case Gelidium agar behaves differently from Gracilaria agar as shown in Figure 14 which illustrates Nikan-Sui gel strength of solutions of agar-carob gum mixtures of 1.5% total colloid concentration. It can be seen that for Gelidium agar, the gel strength increased when substituting part of the agar by carob gum, reaching its maximum strength at an approximate concentration of 1.33% of agar and 0.17% of carob gum. Any further increase of carob gum produces a drop in the gel strength. In the case of Gracilaria agar, the addition of carob gum produces a drop in the gel strength. On the other hand Gracilaria agar used in high sugar concentration solutions (above 50%) increases its gel strength much more than Gelidium agar does.
The data for all these applications refers to industrial agars that are sold worldwide in powder form with different meshes, generally included between 60-100 mesh, ASTM standards, in the Orient there is a considerable household consumption of natural agar with much lower gel strengths, in the range 150-400 g/cm2, with which daily food is prepared at home. This agar is marketed in "strips" or "square" (the appearance is string-like or bar-like respectively, Figure 15) produced always by freezing, thawing, draining, and drying without breaking the strips or squares, that are prepared at the gel stage. This popular method of presentation helps the housewife with her measurements. Such agar, produced basically in Japan, Korea, People's Republic of China and its Taiwan Province, is consumed locally and exported mainly to neighbouring countries with some quantities being sold in Western countries, mainly in health food stores. In Japan the sale of industrial agar for these uses is successfully presented in a pill form of the same content as a bar, to help the housewife with her measurement of it for cooking purposes.
Marketing of industrial agar, food grade, is generally made through trading companies operating from Japan, or in Europe (where the more important commercial centres are located in Hamburg or London), or in the United States where the most important trading companies are located in the areas close to New York.
It is difficult to give an idea of the prices of commercial agar because the usual trade statistics list in the same table, agars with different specifications and applications and therefore with different prices. However taking as a reference the Japanese statistics (since this country produces, Imports, and exports a large quantity of the world agar production) we can see in Figure 16 the export/import prices for 1984-86.
From Figure 16 the average prices for 1984 and 1986 were as follows.
|
Export |
1984 |
1986 |
|
Agar, natural, strip |
US$ 16.17 per kg |
US$ 21.07 per kg |
|
Agar, natural, square |
US$ 28.76 |
US$ 45.61 |
|
Agar, industrial, powder |
US$ 12.76 |
US$ 17.30 |
|
Import |
|
|
|
Agar, natural, strip |
US$ 10.12 |
US$ 13.38 |
|
Agar, industrial, powder |
US$ 12.74 |
US$ 21.40 |
|
Exchange rate |
Yen 246.07/$US |
Yen 154.23/$US |
Due to the large quantities of agar traded by Japan, this data is reasonably representative of the agar world market although the 1986 figures would represent the top market values. This is because the recent strengthening of the yen against most of the major world currencies has allowed some other countries to sell more cheaply.
Part of the industrial agar marketing is handled by companies that trade with food additives as pure products (like agar) or by the preparation of mixtures exclusively formulated for each application.
Figure 14 Agar gel solutions of agar/carob gum
Figure 15 Agar in powder, strip and square forms
Figure 16 Agar imported and exported by Japan in 1984, 1985 and 1986 (January-October)
|
|
|
1984 |
1985 |
1986 (JAN./OCT.) |
|||||
|
QUANTITIES METRIC TONS |
F.O.B. AMOUNT IN YENS |
QUANTITIES METRIC TONS |
F.O.B. AMOUNT IN YENS |
QUANTITIES METRIC TONS |
F.O.B. AMOUNT IN YENS |
||||
|
EXPORTS |
|||||||||
|
I |
NATURAL AGAR |
||||||||
|
|
Strip |
|
7,705 |
30,666,000 |
9,186 |
36,872,000 |
6,664 |
21,651,000 |
|
|
Square |
|
5,779 |
40,894,000 |
5,000 |
35,313,000 |
3,430 |
24,127,000 |
||
|
II |
INDUSTRIAL AGAR |
||||||||
|
|
Powder |
|
866,069 |
2,719,344,000 |
790,248 |
2,666,870,000 |
447,559 |
1,194,436,000 |
|
|
|
TOTALS |
879,553 |
2,790,904,000 |
804,434 |
2,739,055,000 |
457,653 |
1,240,214,000 |
||
|
IMPORTS |
|||||||||
|
I |
NATURAL AGAR |
||||||||
|
|
Strip |
|
119,674 |
298,283,000 |
65,065 |
179,054,000 |
103,078 |
212,693,000 |
|
|
II |
INDUSTRIAL AGAR |
||||||||
|
|
Powder |
|
331,386 |
1,039,658,000 |
420,293 |
1,356,630,000 |
227,231 |
750,114,000 |
|
|
|
TOTALS |
451,060 |
1,337,941,000 |
485,358 |
1,535,684,000 |
330,309 |
962,807,000 |
||
|
EXCHANGE RATE: |
1 US DOLLAR |
YENS 246.07 |
YENS 202.60 |
YENS 154.23 |
|||||
The world market for bacteriological agar is small in relation to the food grade market representing 4-5 % of the total agar sales. A constant and direct contact is established between the technical departments of both manufacturer and consumer, who is in general the manufacturer of dehydrated culture media. Prices are higher than food grade for bulk purchases; they range from US$ 16 to US$ 38 since for some secondary media manufacturing companies a very good grade agar is acceptable but for others with higher standards an agar with distinct specifications is absolutely necessary. However when purchased in one pound jars, the differences between food grade and bacteriological grade become very large since marketing and distribution costs have been added. Researchers buy from this source when they have to formulate their own media. Frequently they use the standard formulations that are produced in large quantities under strict controls by culture media manufacturers, in dehydrated or in ready-to use form, in bottles, tubes or Petri dishes.
A similar situation exists for agarose, the increasing development of which requires a continous contact between both the manufacturer's technical experts and the quality control experts of those companies that distribute biochemical reagents, as well as with the researchers who use agarose in very specialized techniques. Its price is the highest of all polysaccharides derived from seaweed but its market is still small, about 0.2% of the agar market. Prices recorded in commercial catalogues are difficult to compare (for example see Sigma Catalogue for 1987 where thirteen grades of agarose are listed, ranging in price from US$ 535 to US$ 5 400 per kg) as they do not mention the degree to which the corresponding agarose has been modified by synthesis and therefore a range of different products are listed under similar prices categories. So unless a good contact is established with the supplier, purchasing agarose can be confusing for the end user.
There is a small demand for Purified Agar, a special grade of bacteriological agar which can be used in some biochemical applications. Its price lies between US$ 38 and US$ 60/kg, the range being explained by the differences of quality in purified, noble or deionized agars present in the market.
REFERENCES
Alkahane, T. and S. Izumi, 1976. Sulfate groups of the mucilage of red seaweed. Agric.Biol.Chem., 40:285-9
Allan, G.G., et al., 1971. A new procedure for the fractionation of agar. Carbohydr.Res., 17:234-6
Araki, C., 1937. Acetylation of agar-like substance of Gelidium mansii L. J.Chem.Soc.Japan, 58:1338-50
Arnott, S., et al., 1974. The agarose double helix and its function in agarose gel structure. J.Mol.Biol., 90:269-84
Azhitskii, G.Y. and G.C. Kobozev, 1967. Ammonium sulfate use to eliminate first agaropectin and then to precipitate agarose. Lab.Delo, 3:143-5
Barteling, S.J., 1969. A simple method for the preparation of agarose. Clin.Chem., 15:1002-5
Bhattacharjee, S.S., G.K. Hamer and W. Yaphe, 1979. Study of agar and carrageenan by 13C n.m.r. spectroscopy. Proc.Int.Seaweed Symp., 9:379-85
Chueh, C.T. and C.C. Chen, 1982. Seaweed economics in Taiwan. In Proceedings of the cooperative science seminar on cultivation and utilization of economic algae, June, 1978, Guam, University of Guam Marine Laboratory, pp. 9-16, edited by R.T. Tsuda and Y.M. Chiang
Corongiu, G., S.L. Fornili and E. Clementi, 1983. Hydration of agarose double helix: a Monte Carlo simulation. Int.J. Quantum Chem.Quantum Biol.Symp., 10:277-91
Cristiaen, D. and M. Bodard, 1983. Spectroscopie infrarouge de films d'agar de Gracilaria verrucosa (Huds) papefuss. Bot.Mar., 26:425-7
Cross, A.D., 1964. An introduction to practical infrared spectroscopy. London, Butterworth
Di Ninno, V. and E.L. McCandless, 1978. The chemistry and immunochemistry of carrageenan from Eucheuma and related algal species. Carbohydr.Res., 66:85-93
Di Ninno, V. and E.L. McCandless, 1978a. The immunochemistry of lambda-type carrageenan from certain red algae. Carbohydr.Res., 67:235-41
Fuse, T. and F. Goto, 1971. Some properties of agarose and agaropectin isolated from various mucilaginous substances of red seaweeds. Agric.Biol.Chem., 35:799-804
Glickman, S.A. and I.G. Shubtosova, 1957. Physical chemistry of agar theory and practice of agar fractionation. Kolloid Zh., 19:281-6
Glicksman, M., 1983. Red seaweed extracts (agar, carrageenans, furcelleran). In Food hydrocolloids, edited by M. Glicksman. Baton Raton, Florida, CRC Press, pp. 73-113
Hands, S. and S. Peats, 1938. Isolation of an anhydro-L-galactose derivative from agar-agar. Nature, Lond., 142:797
Hirase, S., 1957. Pyruvic acid as a constituent of agar-agar. Mem. Fac.Ind.Arts Kyoto Tech.Univ.Sci.Technol., 1957:17-29
Hirase, S., C.H. Araki and K. Aral, 1968. The synthesis of agarobiose. Bull.Chem.Soc.Japan, 41:626-8
Hjerten, S., 1962. A new method for preparation of agarose for gel electrophoresis. Biochim.Biophys.Acta, 62:445-9
Hjerten, S., 1971. Some new methods for the preparation of agarose. J.Chromatog., 61:73-80
Izumi, K., 1970. A new method for fractionation of agar. Agric.Biol. Chem., 34:1739-40
Jones, W.G.M. and S. Peats, 1942. The constitution of agar. J.Chem. Soc., 1942:225-31
Lahaye, M., C. Rochas and W. Yaphe, 1986. A new procedure for determining the heterogeneity of agar polymers in the cell walls of Gracilaria species. Can.J.Bot., 64:579-85
Letherby, M. and D. Young, 1981. The gelation of agarose. J.Chem. Soc.Faraday Trans.1., 77:1953-66
Liang, J.N., et al., 1979. Spectroscopic origin of conformation-sensitive contributions to polysaccharide optical activity. Vacuum-ultraviolet circular dichroism of agarose. Biopolymers, 18:327-33
Meer, W., 1980. In Handbook of water soluble gums and resins, edited by R.L. Davidson. New York, McGraw-Hill, pp. 7.1 to 7.19
Okazaki, A., 1971. Seaweeds and their uses in Japan. Tokyo, Tokai University Press, pp. 98-161
Patil, N.B. and N.R. Kale, 1973. A simple procedure for the preparation of agarose for gel electrophoresis. Indian J.Biochem.Biophys., 10:160-3
Percival, E.G.V., J.C. Somerville and I.A. Forbes, 1938. Isolation of an anhydro-sugar derivative from agar. Nature, Lond., 142:797-8
Pizarro, A. and H. Barrales, 1986. Field assessment of two methods of planting the agar-containing seaweed, Gracilaria, in northern Chile. Aquaculture, 59:31-44
Rolson, A., 1965. Fractionation of mixtures of agarose and agaropectin. British Patent 1,006,259.
Ren, G.Z. and M.Q. Chen, 1986. The effect of temperature on the growth and development of Gracilaria asiatica. Oceanol. Limnol.Sin., 17:292-300
Ren, G.Z., J.C. Wang and M.Q. Chen, 1984. Cultivation of Gracilaria by means of low rafts. Hydrobiologia, 116/117:72-6
Renn, D.W. 1984. Agar and agarose: indispensable partners in biotechnology. Ind.Eng.Chem.Prod.Res.Dev., 23:17-21
Russell, B., T.H. Mead and A. Polson, 1964. Method of making agarose. Biochim.Biophys.Acta, 96:169-74
Samec, M. and V. Isajevic, 1922. Studien uber Pllanzenkolloide.14. Kolloidchemische, Beihefte 16:5-12
Santelices, B. and R. Ugarte, Production of Chilean Gracilaria: problems and perspectives. In Proceedings of the twelfth International Seaweed Symposium. Sao Paulo, Brazil, August, 1986 (in press).
Selby, H.H., 1954. Agar since 1943. Adv.Chem.Ser.Am.Chem.Soc., 11:16-9
Selby, H.H. and W.H. Wynne, 1973. Agar. In Industrial gums, edited by R.L. Whistler. New York, Academic Press, pp. 29-48
Sigma Chemical Co., 1987. Catalog of biochemical and organic compounds for research and diagnostic chemical reagents. St. Louis, Sigma Chemical Co., pp. 169-70
Stancioff, S.J. and N.F. Stanley, 1969. Infrared and chemical studies on algae polysaccharides. Proc.Int.Seaweed Symp., 6:595-609
Stanley, N.F., 1963. Process for treating a polysaccharide of seaweeds of the Gigartinaceae and Soliesiaceae families. U.S. Patent 3,094,517
Sviridov, S.M., V.A. Berdnikov and V.N. Ivanov, 1971. Agarose isolation from agar. Lab.Delo, 1971:55-7
Tagawa, S., 1966. Separation of agar-agar by dimethyl sulfoxide into agarose and agaropectin. J.Shimonoseki Univ.Fish., 14:165-71
Tagawa, S. and Y. Kojima, 1972. The alkal treatment of the mucilage of Gracilaria verucosa. Proc.Int.Seaweed Symp., 7:447-50
Torres-Pombo, J., 1972. Contribución al conocimiento químico del alga Gelidium sesquipedale (clem) Thuret y a la estructura de su agar. Acta Cient.Compostelana, 9:53-64
Tseng, C.K., 1946. Phycocolloids: useful seaweed polysaccharides. In Colloid chemistry: theoretical and applied, edited by J. Alexander. New York, Reinhold, Vol. 6:629-734
Watase, M. and R. Nishinari, 1983. Rheological properties of agarose gels with different molecular weights. Rheol.Acta, 22:580-7
Yang, S.S., 1982. Seasonal variation of the quality of agar-agar produced in Taiwan. In Proceedings of the Cooperative science seminar on cultivation and utilization of economic algae, June, 1978, edited by R.T. Tsuda and Y.M. Chiang. Guam, University of Guam Marine Laboratory, pp. 65-80
Yaphe, W., 1984. Properties of Gracilaria agars. Hydrobiologia, 116/117:171-86
Zabin, B.A., 1969. Agarose use of DEAE cellulose to remove the anionic polysaccharides from agar. U.S. Patent 3,423,396
BIBLIOGRAPHY
Blanschard, J.M.V. and J.R. Mitchell, 1979. Polysaccharides in food. London, Butterworths
Determan, H., 1969. Chromatographie sur gel. Paris, Masson et Cie
Glicksman, M. (ed.), 1981. Food Hydrocolloids. Vol.1. Comparative properties of hydrocolloids. Baton Raton, CRC Press
Glicksman, M. (ed.), 1983. Food hydrocolloids. Vol.2. Natural plant exudates - seaweed extracts. Baton Raton, CRC Press
International Trade Centre (ITC), 1981. Pilot survey of the world seaweed industry and trade. Geneva, UNCTAD/GATT, International Trade Centre
Levring, T., H.A. Hoppe and O.J. Schmid, 1969. Marine algae. A survey of research and utilization. Bot.Mar.Handb., (1):421 p.
Michanek, G., 1975. Seaweed resources of the ocean. FAO Fish.Tech.Pap., (138)
Percival, E. and R.H. McDowell, 1967. Chemistry and enzymology of marine algal polysaccharides. London, Academic Press
Wieme, R.J., 1965. Agar gel electrophoresis. Amsterdam, Elsevier Publishing Company
INTERNATIONAL SEAWEED SYMPOSIUM PROCEEDINGS
I Black, W.A.P., et al. (eds), 1953. Proceedings of the First international seaweed symposium. Edinburgh, 14-17 July 1952. Inveresk, Midlothian, Scotland, Institute of Seaweed Research for the Organizing Committee, 129 p.
II Braaud, T. and N.A. Sørensen (eds), 1956. Second international seaweed symposium. Trondheim, 14-17 July 1955. London, Pergamon Press, 220 p.
III Heocha, C.O. (ed.), 1958. Galway, Ireland
IV Davy de Virville, A. and J. Feldmann (eds), 1964. Proceedings of the Fourth international seaweed symposium. Biarritz, 1961. London, Pergamon Press, 467 p.
V Young, E.G. and J.L. McLachlan (eds), 1966. Proceedings of the Fifth international seaweed symposium. Halifax, Canada, 25-28 August 1965. Oxford, Pergamon Press, 424 p.
VI Margalef, R. (ed.), 1969. Proceedings of the Sixth international seaweed symposium. Santiago de Compostela, Spain, 9-13 September 1968. Madrid, Subsecretaría de la Marina Mercante, Dirección General de Pesca Marítima, 782 p.
VII Nisizawa, K., et al. (eds), 1972. Proceedings of the Seventh international seaweed symposium. Sapporo, Japan, 8-12 August 1971. Tokyo, University of Tokyo Press, 647 p.
VIII Fogg, G.E. and W.E. Jones (eds), 1981. Proceedings of the Eighth international seaweed symposium. Bangor, North Wales, 18-23 August 1974. Menai Bridge, Wales, U.K., Marine Science Laboratories, 769 p.
IX Jensen, A. and J.R. Stein (eds), 1.979. Proceedings of the Ninth international seaweed symposium, Santa Barbara, California, 20-27 August 1977. Princeton, N.J., Science Press, 634 p.
X Levring, T. (ed.), 1981. Proceedings of the Eleventh seaweed symposium. Göteborg, Sweden, August 11-15, 1980. Berlin, Walter de Gruyter, 780 p.
XI Bird, C.J. and M.A. Ragan (eds), 1984. Proceedings of the Twelfth international seaweed symposium. Quingdao, People's Republic of China, 19-25 June 1983. Dordrecht, Netherlands, Dr W. Junk Publishers, Hydrobiologia, Vol.116/117:624 p.
![]()
![]()
![]()
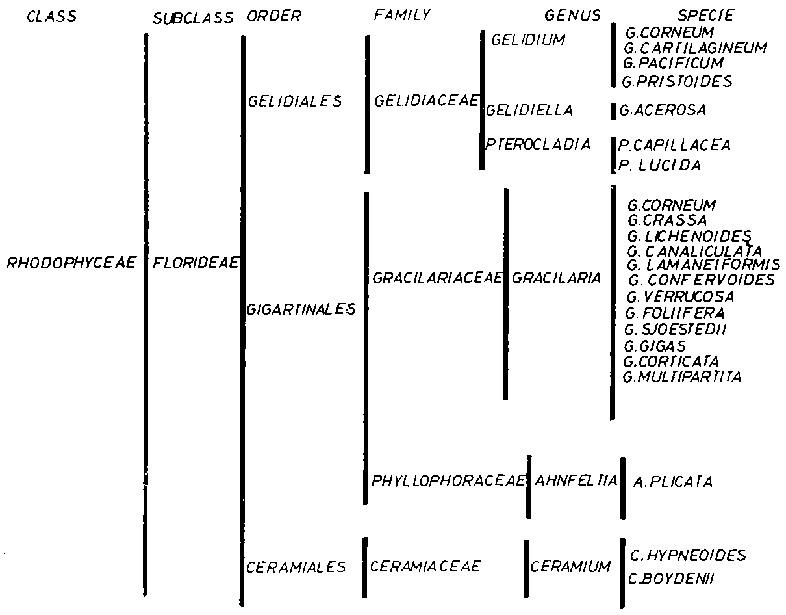{kind=link}
{kind=link}
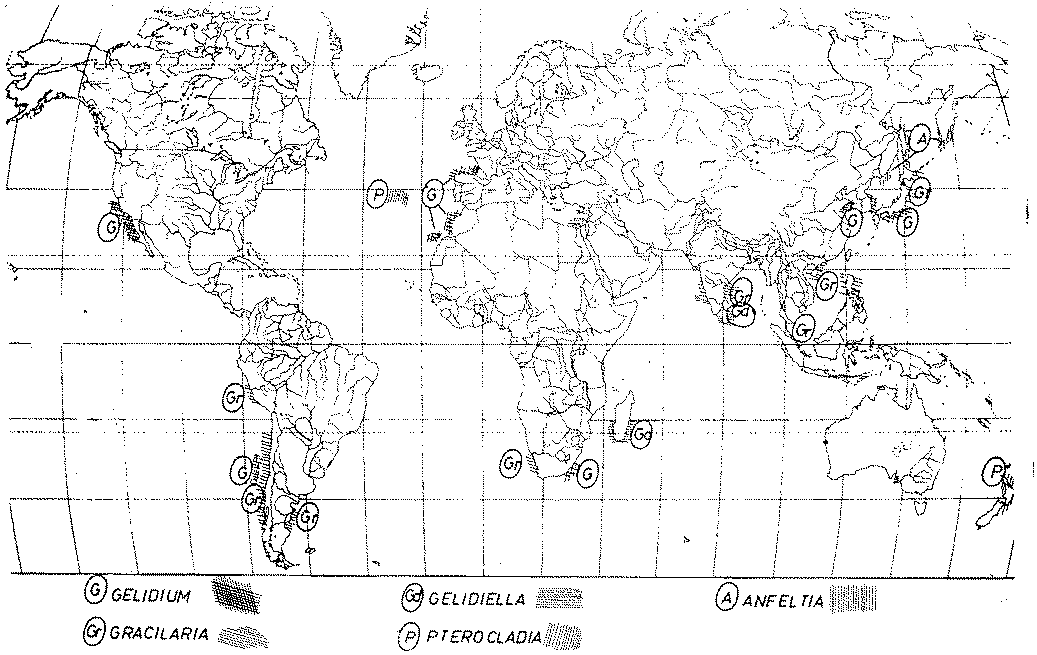{kind=link}
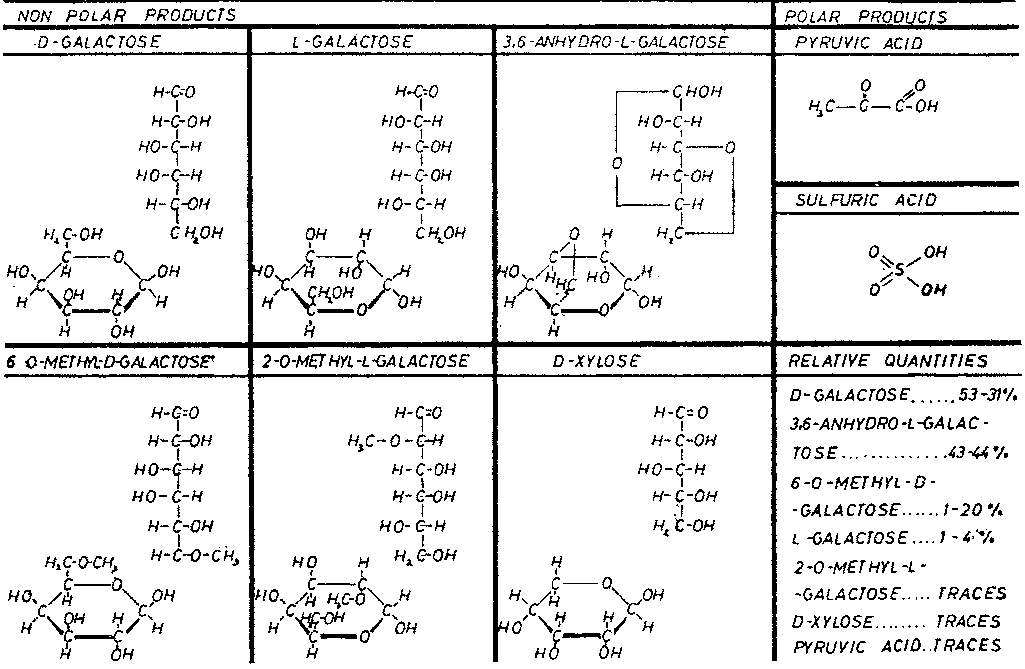{kind=link}
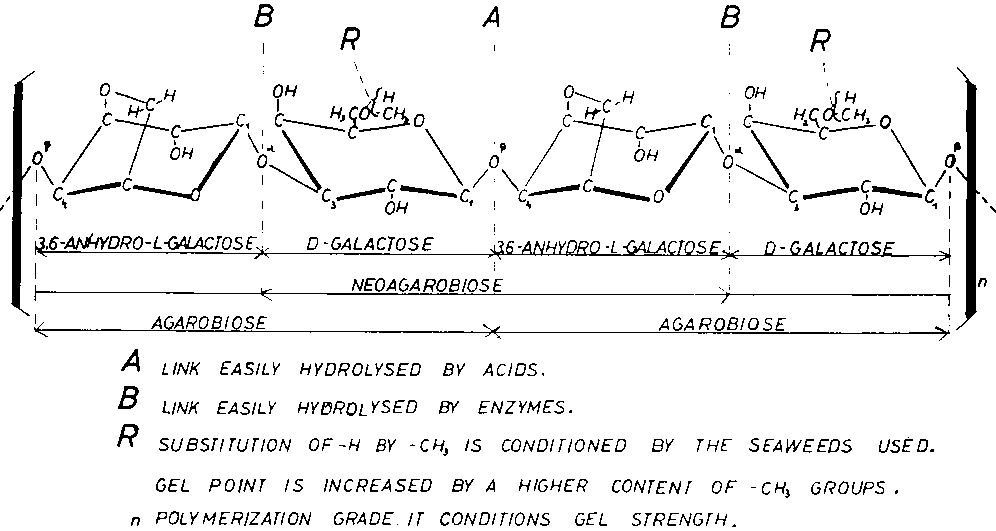{kind=link}
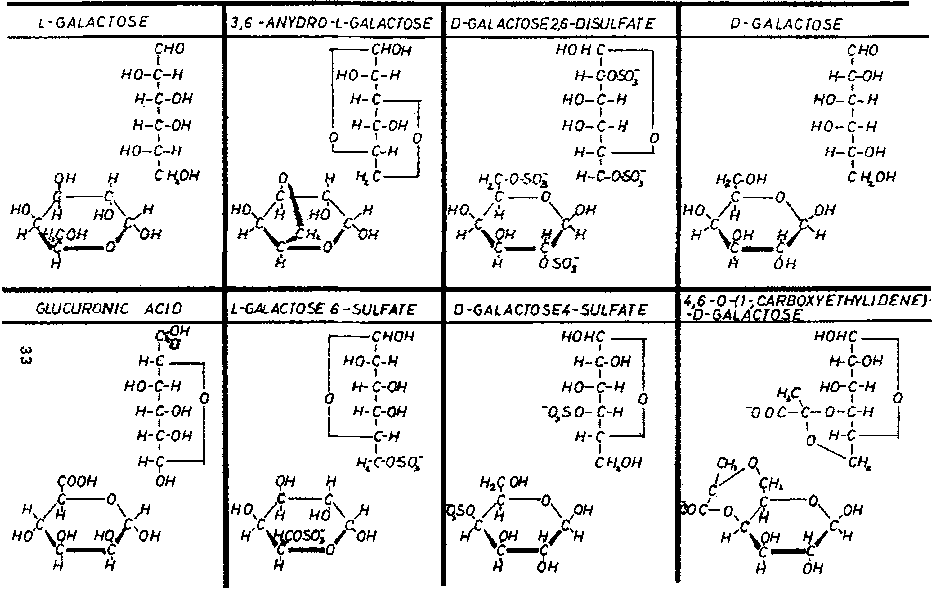{kind=link}
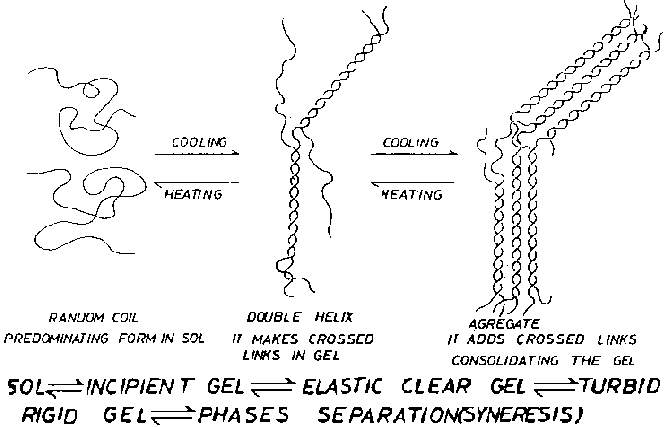{kind=link}
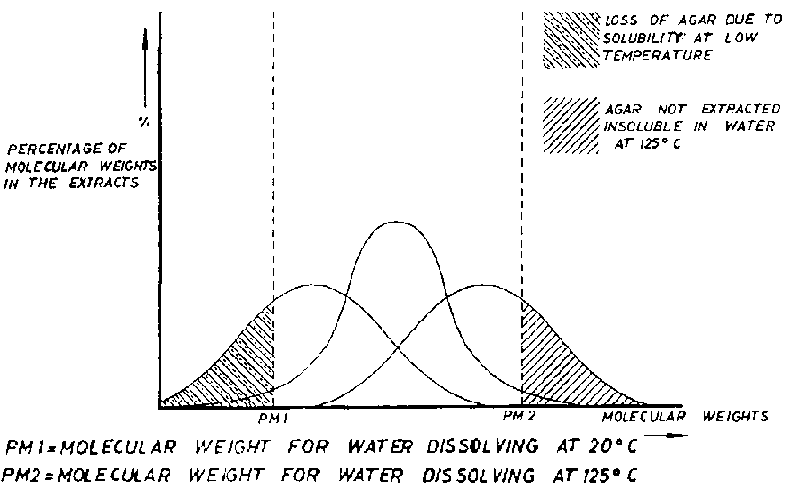{kind=link}
{kind=link}
{kind=link}
{kind=link}