![]()
![]()
![]()
8.1 Introduction
8.2 Rounding and Significant figures
8.3 Control charts
8.4 Preparation of a Control Sample
8.5 Complaints
8.6 Trouble-shooting
8.7 LIMS
SOPs
In the preceding chapters basic elements of quality assurance were discussed. All activities associated with these aspects have one aim: the production of reliable data with a minimum of errors. The present discussion is concerned with activities to verify that a laboratory produces such reliable data consistently. To this end an appropriate programme of quality control (QC) must be implemented. Quality control is the term used to describe the practical steps undertaken to ensure that errors in the analytical data are of a magnitude appropriate for the use to which the data will be put. This means that the (unavoidable) errors made are quantified to enable a decision whether they are of an acceptable magnitude and that unacceptable errors are discovered so that corrective action can be taken and erroneous data are not released. In short, quality control must detect both random and systematic errors.
In principle, quality control for analytical performance consists of two complementary activities: internal QC and external QC.
The internal QC involves the in-house procedures for continuous monitoring of operations and systematic day-to-day checking of the produced data to decide whether these are reliable enough to be released. The procedures primarily monitor the bias of data with the help of control samples and the precision by means of duplicate analyses of test samples and/or of control samples. These activities take place at batch level (second-line control).
The external QC involves reference help from other laboratories and participation in national and/or international interlaboratory sample and data exchange programmes (proficiency testing; third-line control).
The present chapter focuses mainly on the internal QC as this has to be organised by the laboratory itself. External QC, just as indispensable as the internal QC, is dealt with in Chapter 9.
At this point, before entering into actual treatment of data, it might be useful to enter into the data themselves as they are treated and reported. Analytical data, either direct readings (e.g. pH) or results of one or more calculation steps associated with most analytical methods, are often reported with several numbers after the decimal point. In many cases this suggests a higher significance than is warranted by the combination of procedure and test materials. Since clear rules for rounding and for determining the number of significant decimals are available these will be given here.
To allow a better overview and interpretation, to conserve paper (more columns per page), and to simplify subsequent calculations, figures should be rounded up or down leaving out insignificant numbers.
- To produce minimal bias, by convention rounding is done as follows:
- If the last number is 4 or less, retain the preceding number;
- if it is 6 or more, increase the preceding number by 1;
- if the last number is 5, the preceding number is made even.
Examples:
|
pH = |
5.72 |
rounds to |
5.7 |
|
pH = |
5.76 |
rounds to |
5.8 |
|
pH = |
5.75 |
rounds to |
5.8 |
|
pH = |
5.85 |
rounds to |
5.8 |
When calculations and statistics have to be performed, rounding must be done at the end.
Remark: Traditionally, and by most computer calculation programs, when the last number is 5, the preceding number is raised by 1. There is no objection to this practice as long as it causes no disturbing bias, e.g. in surveys of attributes or effects.
8.2.2.1 Rounding of test results
8.2.2.2 Rounding of means and standard deviations
The significance of the numbers of results is a function of the precision of the analytical method. The most practical figures for precision are obtained from the own validation of the procedure whereby the -within-laboratory standard deviation sL (between-batch precision) for control samples is the most realistic parameter for routine procedures (see 7.5.2). For non-routine studies, the sr (within-batch precision) might have to be determined.
To determine which number is still significant, the following rule is applied:
Calculate the upper boundary bt of the rounding interval a using the standard deviation s of the results (n³ 10):
|
bt = ½ s |
(8.1) |
Then choose a equal to the largest decimal unit (...;100; 10; 1; 0.1; 0.01; etc.) which does not exceed the calculated bt
After having done this for each type of analysis at different concentration or intensity levels it will become apparent what the last significant figure or decimal is which may be reported. This exercise has to be repeated regularly but is certainly indicated when a new technique is introduced or when analyses are performed in a nonroutine way or on non-routine test materials.
Example
Table 8-1. A series of repeated CEC determinations (in cmolc/kg) on a control sample, each in a different batch.
|
Data |
Rounded |
|
6.55 |
6.6 |
|
7.01 |
7.0 |
|
7.25 |
7.2 |
|
7.83 |
7.8 |
|
6.95 |
7.0 |
|
7.16 |
7.2 |
|
7.83 |
7.8 |
|
7.05 |
7.0 |
|
6.83 |
6.8 |
|
7.63 |
7.6 |
The standard deviation of this set of (unrounded) data is:
|
|
s =0.4298 |
|
hence: |
bt = 0.2149 |
|
and: |
a = 0.1 |
Therefore, these data should be reported with a maximum of one decimal.
When values for means, standard deviations, and relative standard deviation (RSD and CV) are to be rounded, b is calculated in a different way:
|
for ¯x: |
bx = |
(8.2) |
|
for s: |
bs = |
(8.3) |
|
for RSD: |
brsd = |
(8.4) |
where
¯x = mean of set of n results
s = standard deviation of set of results
RSD = relative standard deviation.
8.3.1 Introduction
8.3.2 Control Chart of the Mean (Mean Chart)
8.3.3 Control Chart of the Range of Duplicates (Range Chart)
8.3.4 Automatic preparation of control charts
As stated in Section 8.1, an internal system for quality control is needed to ensure that valid data continue to be produced. This implies that systematic checks, e.g. per day or per batch, must show that the test results remain reproducible and that the methodology is actually measuring the analyte or attribute in each sample. An excellent and widely used system of such quality control is the application of (Quality) Control Charts. In analytical laboratories such as soil, plant and water laboratories separate control charts can be used for analytical attributes, for instruments and for analysts. Although several types of control charts can be applied, the present discussion will be restricted to the two most usual types:
1. Control Chart of the Mean for the control of bias;
2. Control Chart of the Range of Duplicates for the control of precision.
For the application of quality control charts it is essential that at least Control Samples are available and preferably also (certified) Reference Samples. As the latter are very expensive and, particularly in the case of soil samples, still hard to obtain, laboratories usually have to rely largely on (home-made) control samples. The preparation of control samples is dealt with in Section 8.4.
8.3.2.1 Principle
8.3.2.2 Starting with Mean Charts
8.3.2.3 Using a Mean Chart
In each batch of test samples at least one control sample is analyzed and the result is plotted on the control chart of the attribute and the control sample concerned. The basic construction of this Control Chart of the Mean is presented in Fig. 8-1. (Other names are Mean Chart, x-Chart, Levey-Jennings, or Shewhart Control Chart). This shows the (assumed) relation with the normal distribution of the data around the mean. The interpretation and practical use of control charts is based on a number of rules derived from the probability statistics of the normal distribution. These rules are discussed in 8.3.2.3 below. The basic assumption is that when a control result falls within a distance of 2s from the mean, the system was under control and the results of the batch as a whole can be accepted. A control result beyond the distance of 2s from the mean (the "Warning Limit") signals that something may be wrong or tends to go wrong, while a control result beyond 3s (the "Control Limit" or "Action Limit") indicates that the system was statistically out of control and that the results have to be rejected: the batch has to be repeated after sorting out what went wrong and after correcting the system.
Fig. 8-1. The principle of a Control Chart of the Mean. UCL = Upper Control Limit (or Upper Action Limit). LCL = Lower Control Limit (or Lower Action Limit). UWL = Upper Warning Limit. LWL = Lower Warning Limit.

Apart from test results of control samples, control charts can be used for quite a number of other types of data that need to be controlled on a regular basis, e.g. blanks, recoveries, standard deviations, instrument response. A model for a Mean Chart is given.
Note. The limits at 2s and 3s may be too strict or not strict enough for particular analyses used for particular purposes. A laboratory is free to choose other limits for analyses. Whatever the choice, this should always be identifiable on the control chart (and stated in the SOP or protocol for the use of control charts and consequent actions).
Fig. 8-2. A filled-out control chart of the mean of a control sample.
A control chart can be started when a sufficient number of data of an attribute of the control sample is available (or data of the performance of an analyst in analyzing an attribute, or of the performance of an instrument on an analyte). Since we want the control chart to reflect the actual analytical practice, the data should be collected in the same manner. This is usually done by analyzing a control sample in each batch. Statistically, a sufficient number of data would be 7, but the more data available the better. It is generally recommended to start with at least 10 replicates.
Note: If duplicate determinations of the control sample are used in each batch to control within-batch precision (see 8.3.3), the mean of the duplicates can be used as entry. Although the principle of such a Mean Chart (called ¯x-Chart, as opposed to x-Chart) is the same as for single values, the statistical background of the parameters obviously is not. These two systems may, therefore, not be mixed.
Example
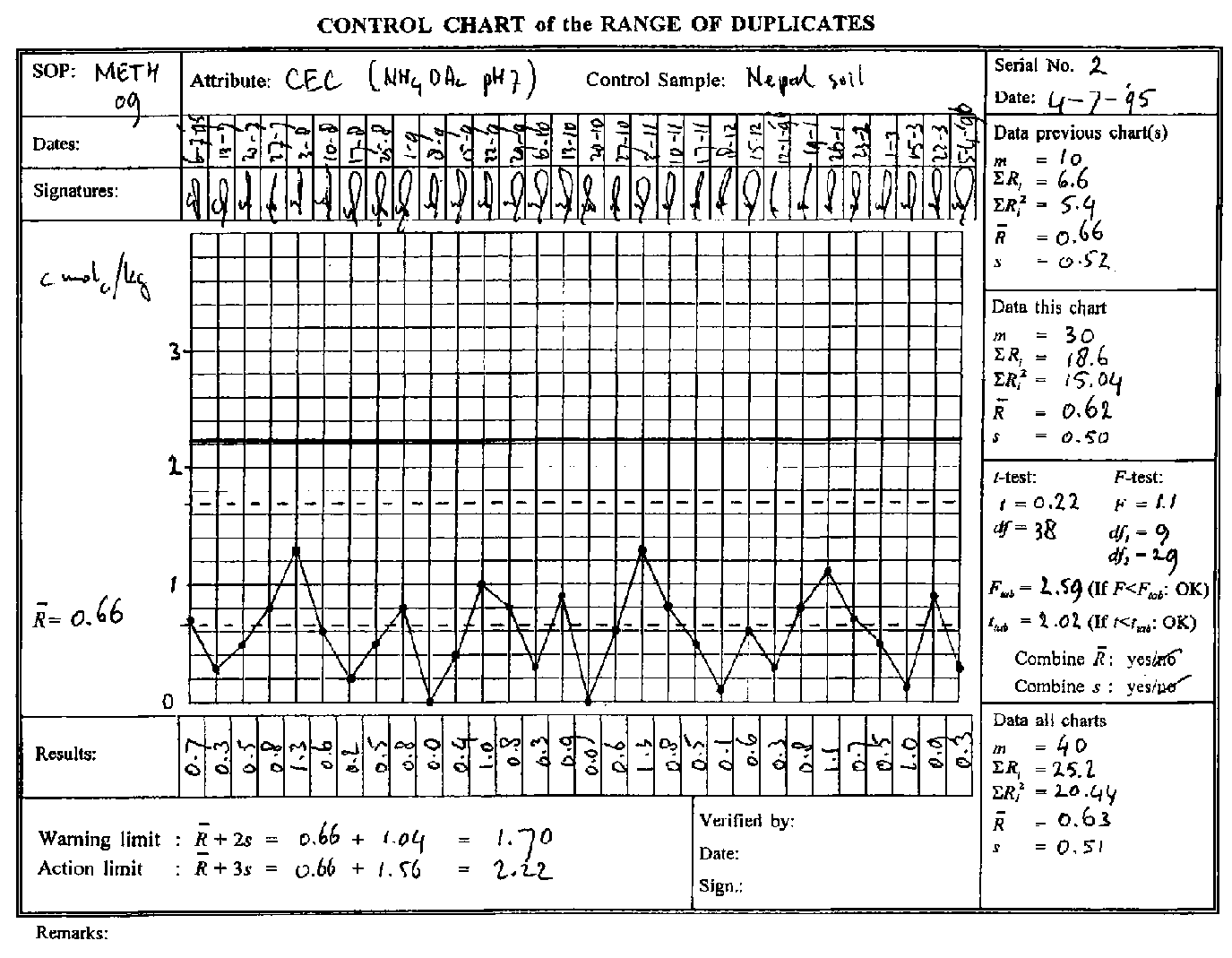
In ten consecutive batches of test samples the CEC of a control sample is determined. The results are: 10.4; 11.6; 10.8; 9.6; 11.2; 11.9; 9.1; 10.4; 10.3; 11.6 cmolc/kg respectively. Using the equations the following parameters for this set of data are obtained: Mean ¯x = 10.7 cmolc/kg, and standard deviation s = 0.91. These are the initial parameters for a new control chart (see Fig. 8-2) and are recorded in the second upper right box of this chart ("data previous chart"). The Mean is drawn as a dashed (nearly) central line. The Warning and Action Limits are calculated in the left lower box, and the corresponding lines drawn as dashed and continuous lines respectively (the Action Line may be drawn in red). The vertical scale is chosen such that the range ¯x ± 3s is roughly 2.5 to 4 cm.
It may turn out, in retrospect, that one (or more) of the initial data lies beyond an initial Action Limit. This result should not have been used for the initial calculations. The calculations then have to be repeated without this result. Therefore, it is advisable to have a few more than ten initial data.
The procedure for starting a control chart should be laid down in a SOP.
After calculating the mean and the standard deviation of the previous chart (or of the initial data set) five lines are drawn on the next control chart: one for the Mean, two Warning Limits and two Action Limits (see Fig. 8-2). Each time a result for the control sample is obtained in a batch of test samples, this result is recorded on the control chart of the attribute concerned. No rules are laid down for the size of a "batch" as this usually depends on the methods and equipment used. Some laboratories use one control sample in every 20 test samples, others use a minimum of 1 in 50.
Note. The level of the analyte in the control sample should as much as possible match the level in the test samples. For this reason it is often necessary to have more than one control sample available for an attribute. To cope with the (expected) variation of the concentration of the analyte in the test samples the use of more than one control sample in a batch must be considered. This would indeed increase the reliability of the obtained results but at a price: an extra analysis is carried out and the chance of false rejection of a batch is increased also.
Quality control rules have been developed to detect excess bias and imprecision as well as shift and drift in the analysis. These rules are used to determine whether or not results of a batch are to be accepted.
Ideally, the quality control rules chosen should provide a high rate of error detection with a low rate of false rejection. The rules for quality control are not uniform: they may vary from laboratory to laboratory, and even within laboratories from analysis to analysis. The rules for the interpretation of quality control charts are not uniform either. Very detailed rules are sometimes applied, particularly when more than one control sample per batch is used. However, it should be realized that stricter rules generally result in (s)lower output of data and higher costs of analysis. The most convenient and commonly applied main rules are the following:
Warning rule (if occurring, then data require farther inspection):
- One control result beyond Warning Limit.
Rejection rules (if occurring, then data are rejected):
- 1. One control result beyond Action Limit.- 2. Two successive control results beyond same Warning Limit.
- 3. Ten successive control results are on the same side of the Mean. (Some laboratories apply six results.)
- 4. Whenever results seem unlikely (plausibility check).
The Warning Rule is exceeded by mere chance in less than 5% of the cases. The chance that the Rejection Rules are violated on purely statistical grounds can be calculated as follows:
|
Rule 1: |
0.3 % |
|
Rule 2: |
0.5×(0.05)2×100% = 0.1% |
|
Rule 3: |
(0.5)10×100% = 0.1% |
Thus, only less than 0.5% of the results will be rejected by mere chance. (This increases to 2% if in Rule 3 'six results on the same side of the mean' is applied.)
If any of the four rejection rules is violated the following actions should be taken:
- Repeat the analysis, if the next point is satisfactory, continue the analysis. If not, then- Investigate the cause of the exceeding.
- Do not use the results of the batch, run, day or period concerned until the cause is trailed. Only use the results if rectification is justified (e.g. when a calculation error was made).
- If no rectification is possible, after elimination of the source of the error, repeat the analysis of the batch(es) concerned. If this next point is satisfactory, the analysis can be continued.
Commonly, outliers are caused by simple errors such as calculation or dilution errors, use of wrong standard solutions or dirty glassware. If there is evidence of such a cause, then this outlier can be put on the chart but may not be used in calculating the statistical parameters of the control chart. These events should be recorded on the chart in the box "Remarks". If the parameters are calculated automatically, the outlier value is not entered.
Rejection Rule 3 may pose a particular problem. If after the 10th successive result on one side of the mean it appears that a systematic error has entered the process, the acceptance of the previous batches has to be reconsidered. If they cannot be corrected they may have to be repeated (if this is still possible: samples may have deteriorated). Also, the customer(s) may have to be informed. Most probably, however, problems of this type are discovered at an earlier stage by other Quality Control tools such as excessive blank readings, the use of independent standard solutions, instrument calibrations, etc. In addition, by consistent inspection of the control chart three or four consecutive control results at the same side of the mean will attract attention and a shift (see below) may already then be suspected.
Rejection Rule 4 is a special case. Unlike the other rules this is a subjective rule based on personal judgement of the analyst and the officer charged with the final screening of the results before release to the customer. Both general and specific knowledge about a sample and the attribute(s) may ring a bell when certain test results are thought to be unexpectedly or impossibly high or low. Also, results may be contradictive, sometimes only noticed by a complaining client. Obviously, much of the success of the application of this rule depends on the available expertise.
Note. A very useful aspect of Quality Control of Data falling under Rejection Rule 4 is the cross-checking of analytical results obtained for one sample (or, sometimes, for a sequence or a group of samples belonging together, e.g. a soil profile or parts of one plant). Certain combinations of data can be considered impossible or highly suspect. For instance, a pH value of 8 and a carbonate content of zero is a highly unlikely combination in soils and should arouse enough suspicion for closer examination and possibly for rejection of either or both results. A number of such contradictions or improbabilities can be built into computer programs and used in automatic cross-checking routines after results are entered into a database. Ideally, these cross-checks are built into a LIMS (Laboratory Information Management System) used by the laboratory. While all LIMSes have options to set ranges within which results of attributes are acceptable, cross-checks of attributes is not a common feature. An example of a LIMS with cross-checks for soil attributes is SOILIMS.
Most models of control charts accommodate 30 entries. When a chart is fall a new chart must be started. On the new chart the parameters of the just completed old chart need to be filled in. This is shown on Fig. 8-2. Calculate the "Data this chart" of the old chart and fill these in on the old chart. Perform the two-sided F-test and t-test (see right, to check if the completed chart agrees with the previous data. If this is the case, calculate "Data all charts" by adding the "Data this chart" to the "Data previous charts". These newly calculated "Data all charts" of the completed old chart are the "Data previous charts" of the new chart. Using these data, the new chart can now be initiated by drawing the new control lines as described in 8.3.2.2.
Shift
In the rare case that the F-test and/or the t-test will not allow the data of a completed control chart to be incorporated in the set of previous data, there is a problem. This has to be resolved before the analysis of the attribute in question can be continued. As indicated above, such a change or shift may have various causes, e.g. introduction of new equipment, instability of the control sample, use of a wrong standard, wrong execution of the method by a substitute analyst. Also, when there is a considerable time interval between batches such a shift may occur (mind the expiry date of reagents!). However, when the control chart is inspected in a proper and consistent manner, usually such errors are discovered before they are revealed by the F and t-test.
Drift
A less conspicuous and therefore perhaps greater danger than incidental errors or shifts is a gradual change in accuracy or precision of the results. An upward or downward trend or drift of the mean or a gradual increase in the standard deviation may be too small to be revealed by the F or t-test but may be substantial over time. Such a drift could be discovered if a control chart were much longer, say some hundreds of observations. A way to imitate this extension of the horizontal scale is to make a "master" control chart with the values of x and s of the normal control charts. Such a compressed control chart could be referred to as "Control Chart of the Trend" and is particularly suitable for a visual inspection of the trend. An upward trend can be suspected in Figure 8-2. Indeed, the mean of the first fifteen entries is 10.59 vs. 10.97 cmolc/kg for the last fifteen entries, implying a relative increase of about 3.5%. This indicates that the further trend has to be watched closely.
The main cause of drift is often instability of the control sample, but other causes such as deterioration of reagents and equipment must taken into account. Whatever the cause, when discovered, it should be traced and rectified. And here too, if necessary, already released results may have to be corrected.
New Control Sample
When a control sample is about to run out, or must be replaced because of instability, or for any other reason, a new control sample must be timely prepared so that it can be run concurrently with the old control sample for some time. This allows to make a smooth start without interrupting the analytical programme. As indicated previously, the more initial data are obtained the better (with a minimum of 10) but ideally a complete control chart should be made.
8.3.3.1 Principle
8.3.3.2 Range chart of Control Sample
8.3.3.3 Starting the first chart
8.3.3.4 R-chart of Test Samples
Between-batch precision (within-laboratory reproducibility, see 7.5.2.3) can be inspected visually on the Control Chart of the Mean; a "noisy" graph with frequent and large fluctuations indicates a lower precision than a smooth graph.
Information about the within-batch precision (repeatability, see 7.5.2.2) can only be obtained by running duplicate analyses in the same batch. For this purpose both test samples and control samples can be used but the latter are somewhat more convenient. The obtained data are plotted on a Control Chart of the Range of Duplicates (also called Range Chart or R-chart).
In each batch of test samples at least one sample is analyzed in duplicate and the difference between the results is plotted on the control chart of the attribute concerned. The basic construction of such a Control Chart of the Range of Duplicates is given in Figure 8-3. It shows similarities with the Control Chart of the Mean in that now a mean of differences is calculated with corresponding standard deviation. The warning line and control line can be drawn at Is and 3s distance from the mean of differences. The graph is single-sided as the lowest observable value of the difference is zero.
Fig. 8-3. Control Chart of the Range of Duplicates. ¯R = mean of the range of duplicates. WL = Warning Limit. CL = Control Limit (or Action Limit).

The simplest way of controlling precision is by running duplicates of a control sample in each batch. The advantage is that this can be directly connected to the use of single values as applied for the Control Chart of the Mean by simply simultaneously running two subsamples of the same control sample. A disadvantage is that precision is measured at one concentration level only (unless more than one control samples are used). The duplicates should be placed at random positions in the batch, not adjacent to each other. The necessary statistical parameters for the Range Chart, ¯R and sR, can be determined as follows:
|
|
(8.5) |
where
¯R = mean difference between duplicates
S Ri = sum of (absolute) differences between duplicates
m == number of pairs of duplicates
and
|
|
(8.6) |
where
sR = standard deviation of the range of all pairs of duplicates.
Fig. 8-4. A filled-out control chart of the range of duplicates of a control sample.
Note 1. Equation (8.6) is equivalent to Equation (7.21). This standard deviation is somewhat different from the common standard deviation of a set of data (Eq. 6.2) and results from pooling the standard deviation of each pair: namely, the duplicates of the pairs have the same population standard deviation.Note 2. If it is decided to routinely run the control sample in duplicate in each batch as described here, a different situation arises with respect to the Mean Chart since now two values for the control sample are obtained instead of one. These values are of equal weight and, therefore, their mean must be used as an entry. It is important to note that the parameters of the thus obtained Mean Chart, particularly the standard deviation, are not the same as those obtained using single values. Hence, these two types should not be mixed up and not be compared by means of the F-test!.
Initiating a Control Chart of the Range of Duplicates is identical to initiating a Control Chart of the Mean as discussed in Section 8.3.2.2. Also the model of the chart is virtually identical with only x replaced by ¯R. The parameters ¯R and sR are determined for at least 10 initial pairs of duplicates as given in Table 8-2 as an example. A control chart with these initial parameters is given in Fig. 8-4.
The interpretation rules of the Range Chart are very similar to those of the Mean Chart:
Warning rule:
- One control observation beyond Warning Limit
Rejection rules:
- One control observation beyond Control (or Action) Limit
- Two successive control observations beyond Warning Limit
- Ten successive control observations beyond ¯R. (Some apply six.)
The response to violation of the rejection rules is also similar: repeat the analysis and investigate the problem if the repeat is not satisfactory.
The procedure to initiate a new chart when the present one is full is identical to that described for the Control Chart of the Mean.
Example
Table 8-2. CEC values (in cmolc/kg) of a control sample determined in duplicate to calculate initial values of ¯R and sR of the control chart of duplicates.
|
|
1 |
2 |
| |
|
|
10.1 |
9.7 |
0.4 | |
|
|
10.7 |
10.2 |
0.5 | |
|
|
10.5 |
11.1 |
0.6 | |
|
|
9.8 |
10.3 |
0.5 | |
|
|
9.0 |
10.1 |
1.1 | |
|
|
11.0 |
10.6 |
0.5 | |
|
|
11.5 |
10.7 |
0.8 | |
|
|
10.9 |
9.5 |
1.4 | |
|
|
8.9 |
9.4 |
0.5 | |
|
|
10.0 |
9.6 |
0.4 | |
|
| ||||
|
Mean: |
10.24 |
10.13 |
¯R: |
0.66 |
|
s: |
0.85 |
0.74 |
sR: |
0.52 |
A limitation of the use of duplicates of a control sample to verify precision is that this may not fully reflect the precision of the analysis of test samples as these may appreciably deviate from the control sample both in matrix and in concentration or capacity of the attribute concerned. The most convenient way to meet this problem is to use more than one control sample with different concentrations of the attribute, each with their own control chart as described above. Another way is to use test samples instead of control samples. However, also in this case duplicates may be chosen at non-representative analyte levels unless the level per batch is rather uniform. Alternatively, all samples are run in duplicate but this is not commonly done in routine analysis and is usually only affordable in special research cases.
When test sample duplicates are preferred two situations can be distinguished:
1. Analyses with a (near-)constant relative standard deviation;
2. Analyses with a non-constant relative standard deviation.
Although commonly occurring, the second case is rather complicated for routine work and will therefore not be treated here.
Constant Relative Standard Deviation
If a constant relative standard deviation (CV or RSD) can be assumed, which may often be the case over certain limited working ranges of concentration, one (or more) test sample(s) in a batch can be analyzed in duplicate instead of a control sample. A constant RSD would result in a control chart as schematically given in Figure 8-5 which is very similar to Fig. 8-3. Because the standard deviation is assumed proportional to the analytical result this applies to the difference between duplicates as well. Therefore, the vertical scale must be the normalized, i.e. the (absolute) value found for R of each pair of duplicates has to be divided by the mean of the two duplicates (and multiplied by 100% if a % scale is used rather than a fraction scale). The interpretation rules and calculations of parameters when a chart is full are again identical to those discussed above for the Control Chart of the Mean.
Fig. 8-5. Control Chart of the Normalized Range of Duplicates. CV = coeff. of variation; other symbols as in Fig. 8-3.

Obviously, in large laboratories with hundreds of analyses per day, much (if not all) of the above discussed control work is usually done automatically by computer. This can be programmed by the laboratory personnel but commercial programs are available which are usually connected to or incorporated in the LIMS (Laboratory Information Management System, see 8.7). For small and medium-sized laboratories (and also for large laboratories starting with control work of new tests or analyses), the manual use of charts, where possible with computerized calculations, is recommended.
8.4.1 Collection and treatment of soil material
8.4.2 Collection and treatment of plant material
8.4.3 Stability
8.4.4 Homogeneity
In the previous sections reference was frequently made to the "Control Sample". It was defined as:
"An in-house reference sample for which one or more property values have been established by the user laboratory, possibly in collaboration with other laboratories."
This is the material a laboratory needs to prepare for second-line (internal) control in each batch and the obtained results of which are plotted on Control Charts. The sample should be sufficiently stable and homogeneous for the properties concerned.
From the foregoing it must have become clear that the control sample has a crucial function in quality control activities. For most analyses a control sample is indispensable. In principle, its place can be taken by a (certified) reference sample, but these are expensive and for many soil and plant analyses not even available. Therefore, laboratories have to prepare control samples themselves or obtain them from other laboratories.
Because the quality control systems rely so heavily on these control samples their preparation should be done with great care so that the samples meet a number of criteria. The main criteria are:
1. The sample is homogeneous2. The material is stable
3. The material has the correct particle size (i.e. passed a prescribed sieve)
4. The relevant information on properties and composition of the matrix, and the concentration of the analyte or attribute concerned is available.
The preparation of a control sample is usually fairly easy and straightforward. As an example it will be described here for a "normal" soil sample (so-called "fine earth") and for a ground plant sample.
Select a location for the collection of suitable and sufficient material. The amount of material to be collected depends on the turn-over of the sample material, the expected stability and the amount that can be handled during preparation. Thus, amounts may range from a few kilos to a hundred kilo or more.
The material is collected in plastic bags and spread out on plastic foil or in large plastic trays in the institute for air-drying (do not expose to direct sunlight; forced drying up to 40°C is permitted). Remove large plant residues. After drying, pass the sample through a 2 mm sieve. Clods, not passing through the sieve are carefully crushed (not ground!) by a pestle and mortar or in a mechanical breaker. Gravel, rock fragments etc. not passing through the sieve are removed and discarded. The material passing through the sieve is collected in a bin or vessel for mechanical homogenization. If the whole sample has to be ground to a finer particle size this can be done at this stage. If only a part has to be ground finer, this should be done after homogenization. Homogenization may be done with a shovel or any other instrument suitable for this purpose. Some laboratories use a concrete mixer. Mixing should be intensive and complete. After that, the bulk sample is divided into subsamples of 0.5 to 1 kg to be used in the laboratory. For this, riffle samplers and sample splitters may be used.
The subsamples can be kept in glass or plastic containers. The latter have the advantage that they are unbreakable. Both have the disadvantage is that fine particles may be electrostatically attracted to the container walls thus causing segregation. The rule about labelling is that it should preferably be done on both the container and the lid. If only one label is used this should always be stuck on the container and not on the lid!
Note. In a note the suggestion is made to have a useful control sample prepared by an interlaboratory sample exchange organization.
Select plant material with the desired or expected composition. Realize that the composition of different parts of a plant (leaf, stem, flower, fruit) may differ considerably and that, in general, the control sample should match the test samples as much as possible.
If the fresh material is contaminated (e.g. by soil, salts, dust) it needs to be washed with tap water or dilute (0.1 M) hydrochloric acid followed by deionized water. For test samples, to minimize the change of concentration of components, this washing should be done in a minimum of time, say within half a minute. For the preparation of a control sample this is less critical.
The sample is dried at 70°C in a ventilated drying oven for 24 hours. The sample is then cut and ground to pass a 1 mm sieve. Storage can be done as described for soil samples.
Note. During the pretreatment (drying, milling, sieving) both soil and plant material may be contaminated by the tools used. In this way the concentration of certain elements (Cu, Fe, Al, etc., see 9.4) may be increased. Like the washing procedure, this problem is less critical for control samples than for test samples (unless the contamination is present as large particles).
No general statement can be given about the stability of the material. Although dried soil and plant material can be kept for a very long time or even, in practice, indefinitely under favourable conditions, it must be realized that some natural attributes may still (slowly) change, that samples for certain analyses may not be dried and that certainly many "foreign" components such as petroleum products, pesticides or other pollutants change with time or disappear at varying unknown rates. Each sample and attribute has to be judged on this aspect individually. Control charts may give useful information about possible changes during storage (trends, shifts).
For quality control it is essential that a control sample is homogeneous so that subsamples used in the batches are "identical". In practice this is impossible (except for solutions), and the requirement can be reduced to the condition that the (sub)samples statistically belong to the same population. This implies a test for homogeneity to prove that the daily-use sample containers (the laboratory control samples) into which the bulk sample was split up represent one and the same sample. This can be done in various ways. A relatively simple procedure is described here.
Check for homogeneity by duplicate analysis
For the check for homogeneity the statistical principles of the two control charts discussed in Section 8.3, i.e. for the Mean and for the Range of Duplicates, are used. The laboratory control samples, prepared by splitting the bulk sample, are analyzed in duplicate in one batch. The analysis used is arbitrary. Usually a rapid, easy and/or cheap analysis suffices. Suitable analyses for soil material are, for example, carbon content, total nitrogen, and loss-on-ignition. For plant samples total nitrogen, phosphorus, or a metal (e.g. Zn) can be used.
The organization of the test is schematically given in Fig. 8-6. As stated before, statistically this test only makes sense when a sufficient number of sample containers are involved (n ³ 7). Do not use too small samples for the analysis as this will adversely affect the representativeness resulting in an unnecessary high standard deviation.
Note. A sample may prove to be homogeneous for one attribute but not for another. Therefore, fundamentally, homogeneity of control samples should be tested with an analysis for each attribute for which the control sample is used. This is done for certified reference samples but is often considered too cumbersome for laboratory control samples. On the other hand, such an effort would have the additional advantage that useful information about the procedure and laboratory performance is obtained (repeatability). Also, such values can be used as initial values of control charts.
Check on the Mean (sample bias)
This is a check to establish if all samples belong to the same population. The means of the duplicates are calculated and treated as single values (xi) for the samples 1 to n. Then, using Equations (6.1) and (6.2), calculate ¯x and s of the data set consisting of the means of duplicates (include all data, i.e. do not exclude outliers).
Fig. 8-6. Scheme for the preparation and homogeneity test of control samples.

The rules for interpretation may vary from one laboratory to another and from one attribute to another. In general, values beyond ± 2s from the mean are considered outliers and rejected. The sample container concerned may be discarded or analyzed again after which the result may well fall within x ± 2s and be accepted or, otherwise, the subsample may now definitely be discarded.
Check on the Range (sample homogeneity)
This is a check to establish if all samples are homogeneous. The differences R between duplicates of each pair are calculated (include all data, i.e. do not exclude outliers). Then calculate ¯R and sR of the data set using Equations (8.5) and (8.6) respectively. The interpretation is identical to that for the Check on the Mean as given in the previous paragraph.
Thus, a laboratory control sample container may have to be discarded on two grounds:
1. because it does not sufficiently represent the level of the attribute in the control sample and
2. because it is internally too heterogeneous.
The preparation of a control sample including a test for homogeneity should be laid down in a SOP.
Example
In Table 8-3 an example is given of a check for homogeneity of a soil control sample of 5 kg which was split into ten equal laboratory control samples of which the loss-on-ignition was determined in duplicate.
The loss-on-ignition can be determined as follows:
1. Weigh approx. 5 g sample into a tared 30 mL porcelain crucible and dry overnight at 105°C.2. Transfer crucible to desiccator to cool; then weigh crucible (accuracy 0.001 g).
3. Place crucibles in furnace and heat at 900°C for 4 hours.
4. Allow furnace to cool to about 100°C, transfer crucible to desiccator to cool, then weigh crucible with residue (accuracy 0.001 g).
Now, the weight loss between 110 and 900°C can be calculated and expressed in mass % or in g/kg (weight basis: material dried at 105 °C).
Table 8-3. Results (in mass/mass %) of duplicate Loss-on-Ignition determinations (A and B) on representative subsamples of ten 500 g laboratory samples of a soil control sample.
|
Sample |
A |
B |
MeanAB |
|
|
1 |
9.10 |
8.42 |
8.760 |
0.68 |
|
2 |
9.65 |
8.66 |
9.155 |
0.99 |
|
3 |
9.63 |
9.18 |
9.405 |
0.45 |
|
4 |
8.65 |
8.89 |
8.770 |
0.24 |
|
5 |
8.71 |
9.19 |
8.950 |
0.48 |
|
6 |
9.14 |
8.93 |
9.040 |
0.22 |
|
7 |
8.71 |
8.97 |
8.840 |
0.26 |
|
8 |
8.59 |
8.78 |
8.685 |
0.19 |
|
9 |
8.86 |
9.12 |
8.990 |
0.26 |
|
10 |
9.04 |
8.75 |
8.895 |
0.29 |
|
Mean: |
8.949 |
0406 |
|
s: |
0.214* |
SR: 0.334** |
(* using Eq. 6.2; ** using Eq. 8.6)
Tolerance range for mean of duplicates (¯x ± 2s):
8.949 ± 2 × 0.214 = 8.52-9.38%
Tolerance range for difference R between duplicates:

In this example it appears that only the mean result of sample no. 3 (= 9.405%) falls outside the permissible range. However, since this is only marginally so (less than 0.3% relative) we may still decide to accept the sample without repeating the analysis.
The measure R for internal homogeneity falls for all samples within the permissible range. (Should an R be found beyond the range we may opt for repeating the duplicate analysis before deciding to discard that sample.)
Errors that escaped detection by the laboratory may be detected or suspected by the customer. Although this particular type of quality control may not be popular, it should in no case be ignored and can sometimes even be useful. For the dealing with complaints a protocol must be made with an accompanying Registration Form with at least the following items:
- name client, and date the complaint was received
- work order number
- description of complaint
- name of person who received the complaint (usually the head of laboratory)
- person charged with investigation
- result of investigation
- name of person(s) who dealt with the complaint
- an evaluation and possible action
- date when report was sent to client
A record of complaints should be kept, the documents involved may be kept in the work order file. The trailing of events (audit trailing) may sometimes not be easy and particularly in such cases the proper registration of all laboratory procedures involved will show to be of great value.
Note. Registration of procedures formally also applies to work that has been put out to contract to other laboratories. When work is put out, the quality standards of the subcontractor should be (demonstrably) satisfactory since the final responsibility towards the client lies with the laboratory that put out the work. If the credibility needs to be verified this is usually done by inserting duplicate and blind samples.
Whenever the quality control detects an error, corrective measures must be taken. As mentioned earlier, the error may be readily recognized as a simple calculation or typing error (decimal point!) which can easily be corrected. If this is not the case, then a systematic investigation must take place. This includes the checking of sample identification, standards, chemicals, pipettes, dispensers, glassware, calibration procedure, and equipment. Standards may be old or wrongly prepared, adjustable pipettes may indicate a wrong volume, glassware may not be cleaned properly, equipment may be dirty (e.g. clogged burner in AAS), or faulty. Particularly electrodes can be a source of error: they may be dirty and their life-time must be observed closely. A pH-electrode may seemingly respond well to calibration buffer solutions but still be faulty.
Clearly, every analytical procedure and instrument has its own characteristic weakness, by experience these become known and it is useful to make a list of such relevant check points for each procedure and adhere this to the corresponding SOP or maintenance logbook if it concerns an instrument. Update this list when a new flaw is discovered.
Trouble-shooting is further discussed in Section 9.4.
8.7.1 Introduction
8.7.2 What is a LIMS?
8.7.3 How to select a LIMS
The various activities in a laboratory produce a large number of data streams which have to be recorded and processed. Some of the main streams are:
- Sample registration
- Desired analytical programme
- Work planning and progress monitoring
- Calibration
- Raw data
- Data processing
- Data quality control
- Reporting
- Invoicing
- Archiving
Each of these aspects requires its own typical paperwork most of which is done with the help of computers. As discussed in previous chapters, it is the responsibility of the laboratory manager to keep track of all aspects and tie them up for proper functioning of the laboratory as a whole. To assist him in this task, the manager will have to develop a working system of records and journals. In laboratories of any appreciable size, but even with more than two analysts, this can be a tedious and error-sensitive job. Consequently, from about 1980, computer programs appeared on the market that could take over much of this work. Subsequently, the capability of Laboratory Information Management Systems (LIMS) has been further developed and their price has increased likewise.
The main benefit of a LIMS is a drastic reduction of the paperwork and improved data recording, leading to higher efficiency and increased quality of reported analytical results. Thus, a LIMS can be a very important tool in Quality Management.
The essential element of a LIMS is a relational database in which laboratory data are logically organized for rapid storage and retrieval. In principle, a LIMS plans, guides and records the passage of a sample through the laboratory, from its registration, through the programme of analyses, the validation of data (acceptance or rejection), before the presentation and/or filing of the analytical results.
Hardware
Originally, LIMSes were installed on mainframe and minicomputers in combination with terminals. However, with the advent of stronger PCs, programs were developed that could run on a single PC (single-user system) or on several PCs with a central one acting as server (network, multi-user system). The more expensive systems allow advanced automation of a laboratory by direct coupling of analytical instruments to the system. Printers are essential parts of the system for label and bar code printing as well as for graphs and reports.
Software
The LIMS software consists of two elements: the routines for the functional parts, and the database. For the latter usually a standard database program is used (e.g. dBase, Oracle,) which can also be done for certain functional parts such as production of graphs and report generation.
The database is subdivided into a static and a dynamic part. The static part comprises the elements that change only little with time such as the definition of analytical methods, whereas the dynamic part relate to clients, samples, planning, and results.
Function features
- A number of common main features of a LIMS are the following:- Registration of samples and assigned jobs with unique numbers and automatic label production.
- Production of work lists for daily and long-term planning.
- Allows rapid insight in status of work (pending jobs, back-log).
- Informs about laboratory productivity (per analysis, whole laboratory).
- Production of control charts and signalling of violation of control rules (results beyond Action Limit, etc.).
- Flagging results beyond preset specifications.
- Generates reports and invoices.
- Archiving facility.
- Allows audit trailing (search for data, errors, etc.).
Data collection and subsequent calculations are usually done "outside" the LIMS. Either with a pocket calculator but more commonly on a PC with a standard type spreadsheet program (such as Lotus 123) or with one supplied with the analytical instrument. The data are then transferred manually or, preferably, by wire or diskette to the LIMS. The larger LIM systems usually have an internal module for this processing.
A major problem with the application of a LIMS is the installation and the involved customizing to the specific needs of a laboratory. One of the first asked questions (after asking for the price) is: 'can I directly connect my equipment to the LIMS?'. Invariably the answer of the vendor is positive but the problems involved are usually concealed or unjustly trivialized. It is not uncommon that installations take more than a year before the systems are operational (not to speak of complete failures), and sometimes the performance falls short of the expectations because the operational complexity was underestimated.
Mentioning these problems is certainly not meant to discourage the purchase of a LIMS. On the contrary, the use of a LIMS in general can be very rewarding. It is rather intended as a warning that the choice for a system must be very carefully considered.
When it is considered that a computerized system might improve the management of the laboratory information data flow, a plan for its procurement must be made. The most important activities prior to the introduction of a
LIMS are the following:
- Set up LIMS project team. Include a senior laboratory technician, the future system manager and someone from the computer department.- Review present procedures and workload.
- Consider if a LIMS can be useful.
Define what the system must do and may cost (make cost/benefit assessment). The cost/benefit assessment is not always straightforward as certain benefits are difficult to assess or to express in money (e.g. improved data quality; changing work attitude). Also, a LIMS may be needed as a training facility for students.
When a decision is made that a LIMS project is viable, the team must define the requirements and consider the two ways to acquire a LIMS: either by in-house building a system or by purchasing one.
Many in-house systems are not premeditated but result from a gradual build-up of small programs written for specific laboratory tasks such as the preparation of work lists or data reports. The advantage is that these programs are fully customized. The disadvantage is that, lacking an initial master plan, they are often not coupled or integrated into an overall system which then takes extra effort. Yet, many laboratories employ such "systems". If a system has to be built from scratch then the general rule is that if a suitable commercial package can be found, it is not economical to build a system as it is both a complicated and time-consuming process.
The purchase of a commercial LIMS should be a well structured exercise, particularly if a large and expensive system is considered. Depending on the capabilities, prices for commercial systems range from roughly USD 25,000 to 100,000 or even higher. The next steps to be taken are:
- Identify LIMS vendors.- Compare requirements with available systems.
- Identify suitable systems and make shortlist of vendors.
- Ask vendors for demonstration and discuss requirements, possible customization, installation problems, training, and after-sales support.
- If possible, contact user(s) of candidate-systems.
After comparing the systems on the shortlist the choice can be made. By way of precaution, it may be wise to start with a "pilot" LIMS, a relatively cheap single-user system in part of the laboratory in order to gain experience and make a more considered decision for a larger system later.
It is essential that all laboratory staff are involved and informed right from the start as a LIMS may be considered meddlesome ('big brother is watching me') possibly arousing a negative attitude. Like Quality Management, the success of a LIMS depends to a large extent on the acceptance by the technical staff.
Remark: Useful information can be obtained from discussions by an active LIMS working group of several hundreds of members on Internet. To subscribe to the (free) mailing list send an E-mail message to: [email protected] stating after Subject: subscribe lims ............ (fill in your name).Another site one may try is: http://www.limsource.com.
|
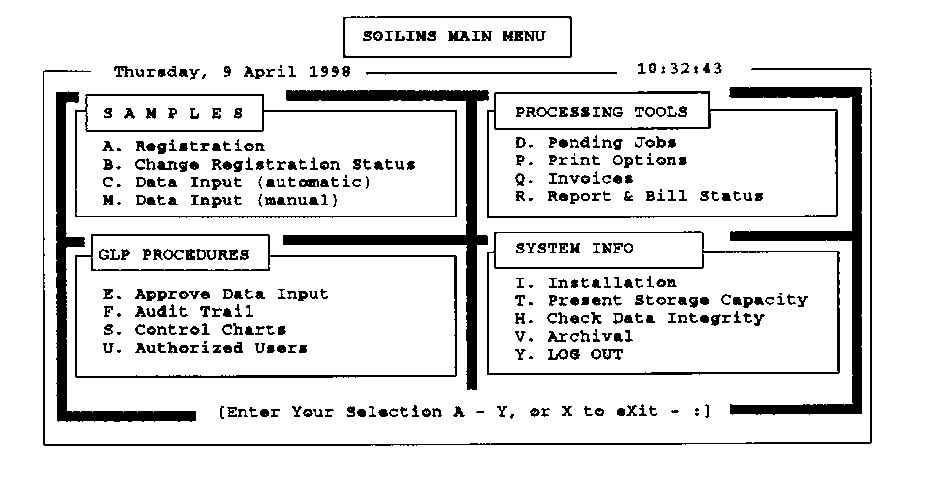
SOILIMS An example of a low-budget and simple stand-alone LIMS specially built for small to medium-sized soil, plant and water laboratories is SOILIMS. It is a user-friendly system which is easily installed and learned (the manual contains a Tutor) and can be used immediately after installation. Although the system has about 100 analyses in the standard configuration, it can be farther customized (and re-customized later) by the supplier. A unique feature is that more than a dozen different cross-checks can automatically be performed in order to screen soil data for internal inconsistencies: when "anomalities" occur, the data concerned are flagged for closer inspection before they are released (anomalities do not necessarily imply errors in all cases). An attractive feature is its price which is comparable to that of a bench-top pH meter. The main features are the following while the system's main menu is given below*. - Unambiguous registration by automatic assignment of unique work order and laboratory sample numbers. - Possibility of priority assignments by deadline definition. - Flexibility to alter work order requests and deadlines. - Time-saving routine for sample label production. - Protection of data against non-authorized users. - Backlog reporting. - Detailed information regarding the status of pending work orders. - Production of work lists provides the manager with complete and accurate information for fast decision making. - Allows for many control samples. - Manual or automatic data input (direct ASCII file reading). - Second-line control by automatic verification of control sample results in Control Charts, - Unique capabilities for cross-checking data ("artificial intelligence"). - Increased efficiency by easy production of reports and invoices. - Data export facilities to LOTUS 123 or text editors. - Easy-to-use automatic archival procedures. - Audit trail capabilities for specified samples, clients, work orders, or laboratory personnel. - Stand-alone and single-user network version. - Option for plant and water analysis included. - Millennium proof. * For more information contact: ISRIC, P.O. Box 353, 6700 AJ Wageningen, the Netherlands. E-mail: [email protected] Minimum required hardware: IBM PC (or compatible) 386 SX with 4Mb RAM. |
Model: Mean Chart
Model: Range Chart
Model: Combined Mean Chart and Range Chart
Model: Combined Mean Chart and Range Chart
![]()
![]()
![]()







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}