Factors affecting the TLC of aflatoxins analysis
Srisit Karunyavanij
Aflatoxins occur in foods and feeds as a result of mold growth and in products such as milk meat and eggs as a result small of ingesting moldy feed by animals. Aflatoxins are potent carcinogens. Their control requires quantitative method of analysis that are sensitive to concentrations of micrograms to picograms per kilogram of food or feed.
Since the first analytical methods for aflatoxins were published in 1964. Thin Layer Chromatography (TLC) has been the only technique cepable of detecting and quantitating aflatoxins at low levels. Now, methods based on higher performance liquid chromatography (HPLC) and radio immunoassay (RIA) or Enzymelinkaged Immuno Sorbent Assay (ELISA) have been developed, but as yet they have not been tested collaboratively.
The aflatoxins are well suited for analysis by TLC since most of the compounds fluoresce strongly under long-wave UV light. Approximately 0.5 ng-spot can be routinely detected either visually or instrumentally. The TLC technique serve as both purification and quantitation step. Before TLC analysis, the aflatoxins are extracted from the sample, usually with an aqueous organic solvent, and the extract is initially purified by one or more techniques such as solvent partition, heavy metal precipitation, column filtration, or chromatography. These techniques affect the result of analysis published by the Association of Official Analytical Chemists (AGAC), the American Oil Chemist Society (AOCS), the European Economic Community (EEC), and the American Association of Cereal Chemists (AACC).
Successful analyses of any commodity depend on the selection of appropriate methods for preparation of extract. Also crucial to successful analysis is thin layer chromatography itself. It is not sufficiently appreciated that good quantitation requires efficient chromatography, i.e., separation of the analysis from each other and from other extractives. Many factors can adversely affect TLC separation and quantitation. These factors are:
THE TLC PLATE
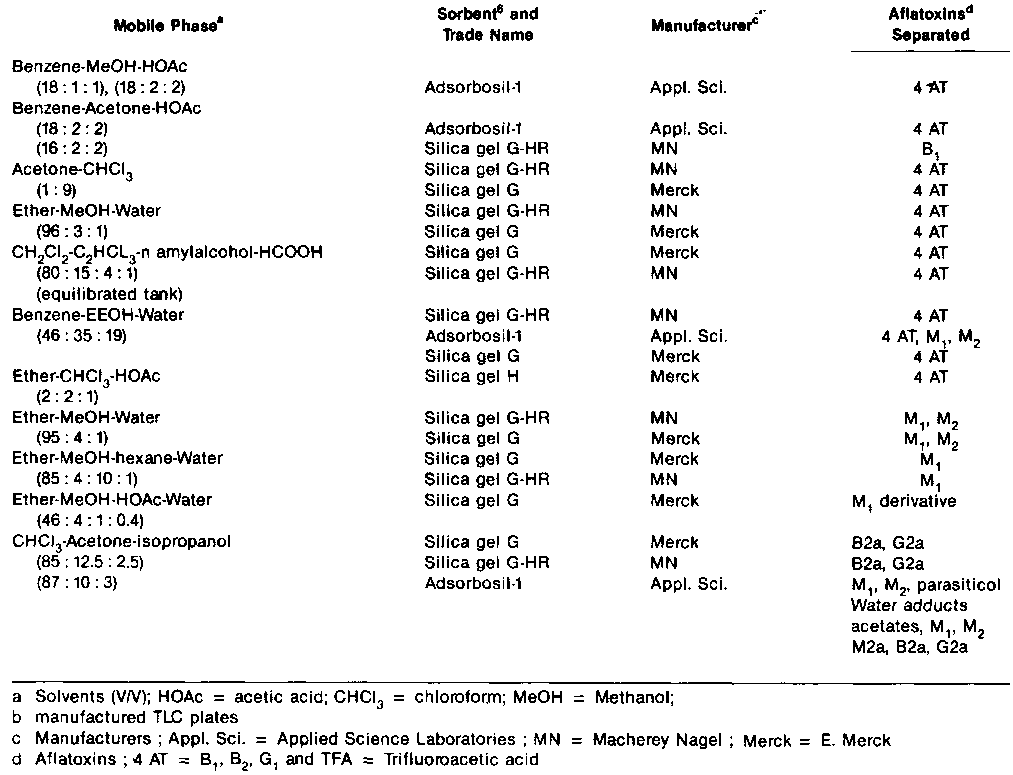
Silica gel is used almost exclusively for TLC of the aflatoxins. The commercially available silica gel vary greatly in their chromatographic properties. These variation show up as difference in their ability to separate the four principal aflatoxins (B1, B2, G1 and G2) from each other and from interference.
Only some of the silica gels completely separate these four toxins with the widely used acetonechloroform as the mobile phase. Some products which have been found satisfactory are list in Table 1, although lot-to-lot differences in their chromatographic properties. The Silica gels also differ in their retentionty for the aflatoxins. Thus with ethyl ether as the developer, migration does not occur on some silica gels but does occur on other. CaSot is the most widely used binder. It produces a soft layer that tends to flake off in the developing solvent. The hard plates produced with polysilicate or organic polymer binders are less prone to mechanic damage, these binders are finding increased use in ready-made or manufacturer prepared plates.
The silica gel can be coated on glass, aluminum or plastic. The advantage of aluminum and plastic is that they can be cut either before or after the chromatography to remove impurities after development or to extract spots for further analysis or identification.
Glass plate is window glass of 0.5 cm thickness and 20x20 cm plate, Glass of this thickness is capable of withstanding handling, clean and heating during repeated use. Thinner glass is too fragile and thicker glass tend to shatter on being heated to 100°C or above. The glass must be clean and free of alkaline residue from cleaning agents, which tend to adhere to glass and which, if left, will cause the aflatoxins to decompose. The alkalirie residues are best removed by rinsing with dilute mineral acid and deimized water.
For small laboratory, laboratory-prepared plates have been preferred: they were cheaper, were generally clean and yielded the best resolution.
Now, commercially prepared TLC plates have improved in uniformity of layer thickness and hardness. They can separate the four aflatoxins (B1, B2, G1 and G2) quite well.
Optimum separation is achieved on plates 0.25 mm thick, sometime the plate up to 2 mm thick have been used for preparatory TLC isolation of the aflatoxin. For best resolution and quantitation, 20x20 cm plates are used but 10x10 cm and 10x10 cm plates have also been used to advantage particularly for preliminary screening. The so called high-performance TLC plate made from a narrow-range silica gel of small particle size in the latest to become available and has been evaluated for aflatoxin.
The hand-make or manufactured TLC plates are activated at 80°-120°c for cat 1 hour. They are stored in a desicator to avoid contamination with atmospheric moisture and pollutants, particularly acids.
TLC plate coating material other than silica gels have been tried but so far have not much use. They include alumine polyamide, formamide-impregnated, diatamaceous earth, or allulose and C18 hydrocarbon bond to TLC silica gel. This system creverse phase TLC of separation on an organic stationary phase using aqueous developing solvents, it works well for the HPLC of aflatoxins.
REFERENCE STANDARDS
Standard aflatoxin are obtained from many sources Table II. The standard is often a source of error in analysis. The accuracy and purity of the standard or standard solution is the responsibility of the individual analyst. The standard solution must be prepared with the same solvent that will be used for the sample extract. Acetonitrite-benzene (2+98) is used for the more soluble aflatoxins (B1, B2, G1 and G2) or Acetonitrite-Benzene (1+9) is used for the aflatoxins to the glass wall of container and (c) detection of deterioration or decomposition during storage of the solution. The aflatoxins are liable to light or UV irradiation both as solid and in solution in such solvents as benzene, chloroform, ethanol methanol, or water. When not used, the standard solutions should be stored in the dark and at low temperature.
SPOTTING OF THE TLC PLATE
The extract is dissolved for convenient application of amounts equivalent to 0.01 9 of original sample for products expected to contain 5-100 ug/kg of aflatoxin B. and extract equivalent to 5 9 of sample for products such as meat, milk and eggs, that are expected to contain less aflatoxin, i.e., between 0.02-10 ug (B1 or M1)/kg. The use of microliter syringe and the application of 1-50 ul of solution per spot has become standard practice in aflatoxin methodology. Application loss than ul without special equipment may give rise to large errors, likewise, the use of large volumes can result in diffused spots and increased chance of damaging the silica gel [ayes.
Application of the sample should be done rapidly under subdued light without pricking or marring the surface, in an atmosphere of less than 60% relative humidity. At higher humidity the silica gel can be protected, (a) by heating the plate during spotting, (b) by covering the silica with a clean glass plate, (c) by spotting under innertgas in a spotting chamber, (d) by spotting the plate after it has been equilibrated with a solvent. The control of moisture is of utmost importance. Some moisture in the silica gel improves the resolution and eliminates tailing. Under very dry conditions, it is necessary to add water to the developing chamber or to the solvent. However, too much moisture results in poor resolution, slow development, and excresively high Rf's. Tecnniques to control humidity also reduce chances that the plate will be contaminanted, such as fluorescent lint or pollutants, that can cause fading of aflatoxins.
For quantitation of the aflatoxin by visual estimation 1, 2, 3 and 5 ul portions of standard solution (1, 1, 0.2, 0.2 ng/ul B1, G1, B2 band G2, respectively) are used. The same volumes of the extract solution are spotted after the aflatoxin concentration in the sample extract has been adjusted to approximately that of standard solution, based on the preliminary TLC analysis. The amount of aflatoxin in the extract is then determined by visually matching the intensity of fluorescene of the sample spots with that of the standard spots. To avoid erroneous identification of unknown spot as aflatoxin, the sample should also be co-chromatographed with superimposed standard aflatoxins (internal standard). This procedure helps overcome the confusion that may arise from uneven development across the TLC plate and from the interaction of aflatoxin with constituents of the extract, particularly fatty materials.
DEVELOPMENT OF THE CHROMATOGRAM
The TLC should be developed as soon as possible with the enosen solvent. Only one chromatogram should be developed at a time in a standard chamber (size 25x25x10 cm). The chamber should be well insulated to minimize temperature gradients (not necessary if the chamber is glass) and sealed to prevent solvent loss. For most solvent combinations the best resolution is obtained in a chamber not equilibrated with solvent before the plate is developed. Equilibration, if used, speeds up the development and gives uniform Rf's across the plate, but at the expense of poorer resolution. Development (10-12 cm) should take 30-90 minutes depending on the solvent, the silica gel particle size, and activity (moisture content). A fixed volume of freshly mixed solvent should be used and preferably placed in a solvent trough rather than in the bottom of the chamber. This procedure saves solvent and improves reproducibility and resolution of the chromatography.
MOBILE PHASES
In choosing solvents and solvent combinations the analyst can take advantage of the wide range of solvent selectives for the individual aflatoxins and interfering constituents of the extract and greatly improve the analytical results. Tables I and III list many of the mobile phases that have been used. The neutral acetonecnioroform mixture is recommended for testing the performance of silica gels and other conditions used for TLC. The acidic developers Benzene-acetic acidmethanol give excellent resolution of aflatoxins B1, B2, G1 and G2 from each other and from certain extract interferences, even though many acidic and most other materials are moved to higher Rf's relative to the aflatoxins than they are with acetone-chloroform. Benzene-ethanol-water and ether-methanol-water are examples of developers in which the separation may take place by partition. The moisture content of the silica gel has less of an effect on separation with aqueous solvents than with mixtures containing no water.
QUANTITATION
The estimation of aflatoxins visually or by densitometry by measuring the intensity of fluorescence of the aflatoxin spots is the most widely used technique. Basically, in visual estimation two techniques have been used. One is the method of serial dilution of unknown extracts and standard to the point at which fluorescence is not detectable (about 0.1-0.5 ng for aflatoxin B. or G.). This point of extinction depends on the intensity of the UV light, the silica gel, and the residual solvent in the silica gel, as well as the compactness of the spot, the quantity and type of extract interferences, the darkness of the room and the visual acuity of the observer.
The second technique for visual estimation involves comparing the fluorescence intensity of extract spots with those of standard aflatoxin spots. Differences of abound 20% are detectable in the range of 0.2-10 ng aflatoxin. For viewing the TLC spots a Chromato Vue cabinet equipped with one or more 15W, longwave UV lights is adequate. The eyes should be allowed to adjust to subdued lights bofore the fluorescence intensity comparisons are made. If the intensity of the unknown spot is between those of two of the standard spots it is interpolated as half the difference of the volume or amounts in the two spots, i.e., 2.5 if between 2 and 3 ug ng. If the sample falls outside the range of the standard spots, it must be diluted or concentrated and rechromatographed; estimates must never be made by extrapolation from a series of the standard spots.
Densitometry by UV absorptions spectophotometry has been used but because it requires microgram-amount of toxin for detection, it is not practical for analysis of samples in the ug/kg range. Fluoredensitometry is the most widely used technique and is gradually supplanting visual estimation. Again, good chromatography with well-resolved, small spots is essential for good quantitation, even more than for visual estimation, and the same factors influence both humidity, silica gel, developing solvent, solvent residues, layer thickness and uniformity, contaminants, Rf, and type and mode of operation of instrument.
One of the basic problems in the instrumental measurement of the aflatoxin spots is minimizing the effect of extract interferences, a problem as yet only partially solved. One approach is that of standard addition, i.e., graded amounts of standard aflatoxin are added to the unknown extract. The observed responses are plotted against amounts added. The amount of aflatoxin originally present in the unknown is calculated from the observed response for the point at which no aflatoxin was added. This technique is more tedious than others and offers no compensating advantages. The second approach is to spot the standard at four concentrations, close to dried including the concentrations of the unknowns, construct a standard curve for each TLC plate, and make the determination of the unknown from the standard curve. This would appear to be the safest approach because the system is tested for linearity for each chromatogram. The third approach as described in AOAC method is the simplest, most efficient, and most widely used. The sample and standards are then spotted in duplicate at similar concentrations. In this procedue only an occasional check on the response linearity is done as an analytical quality control.
In the scanning of the chromatogram the following precautions may help improve the results, Because the amount and nature of the solvent remaining in the silica affects the fluorescence intensity of the aflatoxins, volatile developing solvent must be rigorously eliminated. For the solvents commonly used drying the plate 15 minutes at 25°C or 5 minutes at 40-80°C is sufficient. Fading of the aflatoxins can be prevented by covering the chromatogram while still hot with a clean glass plate. Although it is advisable to scan the plate after development as soon as possible, some delay can be tolerated. If delay is necessary, the chromatograms are best stored in the dark, protected from atmospheric reactants, prefesably in a cold place. Storage for as long as a week has shown no problems.
The spots should be scanned in direction parallel to the direction of development and from low to high background. The duration and intensity of the UV and other light exposure must be kept to a minimum to avoid undesirable changes. Exposure of aflatoxin to light for 4 hours has resulted in a 40% reduction. If fading of the aflatoxin spots occurs, the standards usually fade faster than the aflatoxin spots in extract. The standards should therefore be scanned before the unknowns. The TLC and scanning results should be evaluated as they are being carried out. Duplicate spots should agree within ± 5% or else be rescanned.
The instrumental parameters to be optimized include the following: selecting excitation light filters or monochromator setting for maximum response; setting the gain for maximum signal - to noise ratio; selecting scanning and recorder speeds: selecting UV cutoff filter or monochromator setting before the detector for maximum response, minimum noise, and case and accuracy in measurement of instrument response; and selecting optimum slit widths and lengths for the best signal-to-noise ratio consistent with adequate resolution of adjacent spots. The output from the instrument can be a strip chart recording on which peak areas can be measured manually or mechanically, it can be electronically integrated using any of many types of integrators.
After the appropriate instrumental parameters are selected, the linearity of response to concentration must be determined by preparing a standard curve covering the range of concentrations of interest. For instruments such as the Schoeffel, peak areas appear best related to concentration, whereas the Zeiss spectrophotofluoro-densitometer accurately measures concentration by peak height as well.
MULTIPLE DEVELOPMENT AND TWO-DIMENSIONAL TLC
For many commodities, i.e., eggs, cheeses, tissue milk figs, spices, mixed feeds and fish-meals, one-dimensional TLC is not is not adequate for separating the aflatoxins from interfering constituents. It is also not adequate when levels lower than 1 ug/kg are to be detected.
The simplest technique for improving separation is multiple development with the same or different solvents (Table III). An extension of this technique is a procedure in which the sample and standard are spotted along a horizontal line midway on a silica gel coated alumina sheet. The sheet is predeveloped with ethyl ether which moves many interferences to the top of the chromatogram but leaves the aflatoxins near the origin. The top of the chromatogram is cut off and the sheet is rotated 180° and developed with a solvent appropiate for aflatoxins. An apparatus that automatically performs multiple development is commercially available. It has been applied to aflatoxin in a preliminary test.
The technique of two-dimensional is timeconsuming, because only one sample can be applied perTLC plate. Usually the standards are developed in only one direction, but this appears to be satisfactory as demonstated in studies room. Both visual methods and densitometry can be used for estimation.
CONFIRMATION OF IDENTITY
Because initial identification of unknowns in a chromatogram is based on the similarities of their Rf values with those of standard aflatoxins, additional proof of identification is needed. This need can hardly be over-emphasized, particularly for regulatory samples and for commodities with a limited history of analysis or incidence of contamination. Several techniques that have been used for confirmation of identity (Table IV). Rechromatography with several different solvent such as in Table 1 and the use of the spray reagents in Table IV are not specific tests and do not give conclusive positive identification. They are conclusive only when they show that the unknown is not aflatoxin. The mineral acid sprays are widely used tests; the acid changes the fluorescence of the aflatoxins from blue to yellow. If an unknown remains blue fluorescent on spraying, one can safely conclude that it is not aflatoxin.
The preparation of chemical derivatives of the aflatoxins with changed chromatographic properties is the simplest method for positive confirmation of chemical identity. Widely used tests for aflatoxin B., G. and M, involve the formation of a water addition product, with trifluoroacetic acid as the catalyst, or formation of acetates. TLC derivatives of the unknowns are then compared by TLC with the derivatives of standard aflatoxins, the derivative are formed directly on the TLC either before or after it is developed.
Move specific than any to the above techniques is that of mass spectrometry. Recent improvements have greatly lowered its detection limits, now approaching nanogram amounts of aflatoxin B1 from crude extracts.
Table 1. Mobile Phase and Sorbents for Thin Layer Chromatography of Aflatoxins
Table II Source of Mycotoxin Reference Standard
—————————————————————————————————————————
—————————————————————————————————————————
DETECTION, ESTIMATION AND CONFIRMATION OF AFLATOXIN BY THIN LAYER CHROMATOGRAPHY (TLC)
Several TLC methods may be used to estimate the concentration of aflatoxins.
The method routinely used at TDRI is the "comparison-of-standards" technique, where known concentrations of pure aflatoxin standards are chromatographed alongside portions of the extract. The fluorescent intensities of the spots on the developed chromatograms are compared visually or instrumentally. This method has superseded the "dilution-to-extinction" technique where the minimum amount of aflatoxin required to produce a fluorescence is known, and used as a standard factor and the sample is diluted until the extinction point is reached.
APPLICATION OF EXTRACTS ONTO TLC PLATES
The method is the same for all TLC techniques. Apply the solutions to the chromatoplate using disposable micro-pipettes, renewing the micropipettes after application of each solution. The extracts should be applied such that the resultant spots are as compact as possible (less than 5 mm diameter). The volumes of extracts and standards applied to the plates vary according to the method of estimating and suspected concentration of the extract.
UNI-DIMENSIONAL CHROMATOGRAPHY
Development of Plates
Develop the plate by standing it in a chromatography tank containing the appropriate solvent to a depth of not more than 0.5 cm. For some solvent systems, the chromatography tank should be left for a short time to equilibrate before the plates are developed. The development time is approximately 20 minutes, depending on the environmental conditions, (the solvent - front must be at least 10 cm from the baseline). Examine the dry, developed plate under long wave (365 nm) ultra violet light, preferably in an enclosed viewing cabinet fitted with a protective filter.
Detection and Estimation
(a) "Comparison of standards" technique
Compare the fluorescence intensities of the spots at the Rf of B1 in the sample with those of the B1 standard spots and determine which of the sample spots matches one of the standards and record the corresponding aliquot volumes. If the sample spot intensity lies between two adjacent standard spots the average liquor volume of the standard spots is recorded. If the spots of the smallest volume of sample are too intense to match the standards the sample extract should be diluted and re-chromatographed.
The concentration of aflatoxin B1 in the sample in ug/kg, is calculated from the formula:
Aflatoxin B. content (ug/kg) = ![]()
Where
S = Volume, in ul of aflatoxin B1
standard of equivalent intensity to Z ul of sample.
Y = concentration of aflatoxin B1 standard in ug/ml
Z = Volume, in ul, of sample extract required, to give
fluorescence intensity comparable to that of S ul of the B.
standard.
V = Volume, in ul, of solvent required to dilute final extract.
W = Weight, in g, of original sample contained in final extract.
At TDRI this is termed, final “effective weight".
Proof of the equation ![]()
From the TLC plate:
S ul standard = Z ul sample
The weight of aflatoxin in the sample (Z ul) and standard (S ul) spots are equal.
Weight of aflatoxin in the standard spot: (S ul)
The concentration of standard
= Y ug/ml or ![]() ug/ul
ug/ul
Therefore weight of aflatoxin is S ul
![]()
Weight of aflatoxin in sample spot: (Z ul)
This must also ![]()
Therefore Z ul sample contains ![]() aflatoxin
aflatoxin
The V ul final sample extract
must contain ![]() ug and this is the weight of aflatoxin in
the final "effective weight" (W) of starting material.
ug and this is the weight of aflatoxin in
the final "effective weight" (W) of starting material.
Thus the weight of aflatoxin in 19 starting material
![]()
Therefore weight of aflatoxin in 1 kg of starting material
![]()
Example using "Comparison -fo - Standards" methods of estimation
20 g comminuted groundnuts were defatted to give 10.9 g material, 10 g of which was extracted with 100 ml chloroform and 10 ml water. The extract was filtered and 50 ml extract was dried and made up to 0.5 ml then, 3 ul 5 ul and 10 ul of this solution were spotted onto a chromatoplate together with 2 ul, 5 ul and 10 ul of standard B1 solution. After development, inspection of the chromatoplate under UV light showed that the 5 ul spot of the extract was equal in intensity to the 2 ul standard spot.
Using the equation: ![]() ug/kg (ppb) - described above.
ug/kg (ppb) - described above.
In this case:
S = 2 ul
Y = 0.66 ug/ml
V = 0.5 ml or 500 ul
Z = 5 ul
W = 9.18 g
The "Effective Weight" (W) for this example is calculated es follows:
20 g sample yielded 10.9 g defatted material. (It is assumed that all the aflatoxin remains in the meal).
Thus the "effective
weight" at this point = ![]() starting
material
starting
material
All the aflatoxin extracted from the 10 g sample will be homogeneously distributed in the 100 ml of chloroform. The 10 ml of water used in the extraction is absorbed by the sample and is therefore disregarded.
The "effective weight" (18.35 9) at this point is, therefore, contained in 100 ml of CHC13.
But after filtration, only 50 ml
filtrate is used for further analysis and the "effective
weight" at this point is therefore given by ![]() of starting material
of starting material
The 50 ml of extracted material is concentrated to 0.5 ml and, contains all the aflatoxins which were present in the 50 ml volume. Therefore, the "effective weight" does not change during the concentration step.
Therefore the aflatoxin content of the sample
![]()
(b) "Dilution-to-extinction" technique
Under UV (365 nm) light look for a blue fluorescence in the sample at an Rf corresponding to aflatoxin B (B1 and B2 will not be resolved) in the standard reference marker. The object of this technique is to find a dilution of the extract, such that, a 15 ul or 20 ul aliquot applied to the TLC plate will give a blue fluorescence of aflatoxin B which is JUST VISIBLE. This means that the 5 ul and 10 ul aliquot will give no fluorescence but in the 25 ul aliquot the fluorescence will be easily visible. Consequently, if the chromatogram shows blue fluorescences in all the aliquots, then the extract has to be diluted sufficiently to give the above extinction point. Trial and error and eventually experience enables the correct dilution to be made and the extinction point determined efficiently.
It is essential that the conditions used for this method are standard, ie:
Under these conditions the smallest weight of aflatoxin B giving a fluorescence which is just visible is 0.4 ng (0.0004 ug), and for aflatoxin G is 0.3 ng (0.0003 ug).
This factor is used in calculating the aflatoxin content as follows,
![]()
![]()
![]()
![]()
Where
D = total volume, in ml,
necessary to dilute the extract so that the fluorescence is just
visible in V ul of sample extract
V = aliquot volume, in ul, at which the fluorescince is just
observed on the developed chromatoplate
W = weight, in g, of original sample contained in the extract
Proof of the above equations:-
A "just visible" fluorescence at Rf B (or G) in the sample extract contains 0.4 ng aflatoxin B (or 0.3 ng aflatoxin G).
This was applied to the plate in V uls sample extract, taken from a final dilution volume of D mist
Thus if V uls aliquot contains 0.4 ng (or 0.3 ng) aflatoxin
D ml (D x 1000 ul) will contain ![]()
aflatoxin B
D ml also contains the "effective weight" (W), so weight of aflatoxin in Wg is:
![]()
Therefore I g of starting material contains
![]()
and 1 kg of starting material contains
![]()
Example using dilution to extinction method of estimation
Assume that the defatting, extraction and filtration procedures are the same as in the sample on page 138. Take 50 ml filtrate and concentrate this to 5 ml. Spot 5, 10, 15, 20 and 25 ul volumes on to a Kieselgel "G" chromatoplate. After development of the plate the fluorescent spot of aflatoxin B is just visible (under the prescribed conditions of illumination) in the 15 ul volume but not in the 10 or 5 ul volumes.
For this example the calculation is as follows:
![]()
or
![]()
Where
D = 5 ml
V = 15 ul
W = 9.18 g (calculating of the effective weight is as shown in the previous example).
Therefore: ![]()
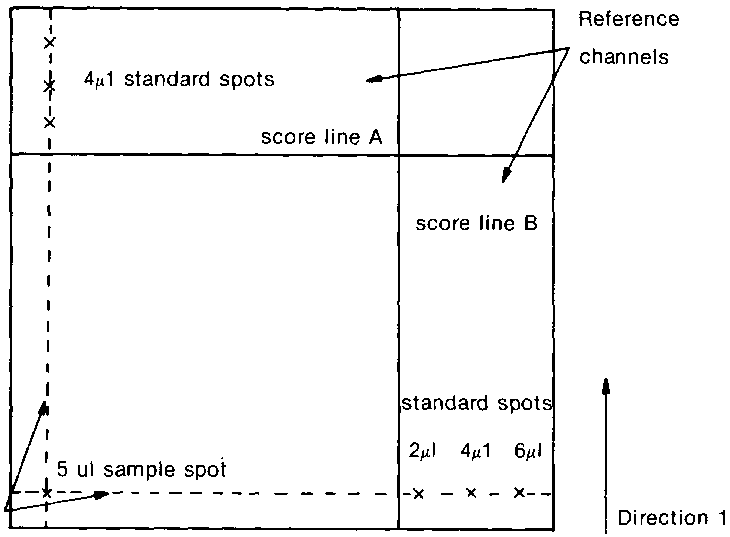
TWO DIMENSIONAL TLC
Two-dimensional TLC can provide a means of incorporating a "clean-up" step into the quantification procedure ("comparison-of-standards" technique) of the analysis.
Standards are applied to the plate on the pencil lines in the reference channels and the sample is applied at the junction of these two lines (see diagram above). This means that the sample is in line with the standard when the plate is developed in both directions. Only one sample can be assessed at a time. The same solvent system can be used in both directions, or two different systems can be used. Another plate is scored and spotted, identical to that just spotted, with the exception that an extra 5 ul of standard solution is applied on top of the sample spot, which acts as an internal marker for the toxin on this so-called "spiked" plate. The former "unspiked" plate is developed in the same tank as the "spiked" plate, so that the development conditions are exactly the same for both plates. The plates are placed in the tanks such that their coated surfaces face towards the centre, but do not touch. The solvent is allowed to rise up the plate until score line A is reached. The plate is removed and thoroughly dried in the dark (no solvent residue should remain).
Diagram showing chromatograms and area of plate affected by solvent after development in direction 1
The plate is turned through 90° and developed in the second solvent system
After development in second direction
Final chromatoplate (viewed as spotted) showing pathway of the toxin fluorescences
It can be seen that the Rf values of the toxin fluorescence in the "sample" and "spiked-sample" do not correspond exactly to the standard Rf values in the reference channels but are slightly further advanced in direction (2) towards the right of the plate. This is because the coated surface of the plate within the score lines A and B is deactivated by the first developing solvent prior to the second development whereas the coating in the reference channels is not affected. Obviously these differences will depend on solvent systems and coating material used.
INSTRUMENTAL EVALUATION OF TLC PLATES
The methods described above for estimating the concentration of the mycotoxin extract, either by "dilution-to-extinction" or by "camparison-of-standards" techniques, are subjective measurements. Due to the difficulty of estimating small differences in fluorescent intensity with the eye, visual estimating has a coefficient of variation of about 30% even under ideal conditions. These limits of precision are, however, sufficient for most purposes.
A number of reports have appeared which describe the use of any one of a number densitometers available for measuring the fluorescent intensity of mycotoxins on TLC plates. The results indicate that both the accuracy and provision of mycotoxin analysis can sometimes be enhanced when objective instrumental measurements of the intensity of the fluorescence are made, but this is influenced by factors such as errors in spotting, poor resolution with some batches of silica gel etc.
Collaborative studies have indicated that the lack of reproducibility arising from these factors negates any advantage obtained by utilising the more accurate detector within the densitometer. A coefficient of variation of 30% was obtained irrespective of whether visual or densitometric estimation was employed.
CONFIRMATION
After the identification and estimation steps of the analytical procedure, it is ESSENTIAL to include an additional step which confirms, unambiguously, the authenticity of the mycotoxins. There are a number of confirmatory procedures available and many are based on the formation of coloured or fluorescent mycotoxin derivatives. The following methods are those used routinely at TDRI for confirming the presence of the aflatoxins.
SAFETY PRECAUTIONS
Laboratory Facilities Required
Precautions During Analysis
Precautions During TLC
REFERENCES
Anon. (1980) Laboratory Decontamination and Destruction of Aflatoxins B1, B2, G1, G2 in Laboratory Wastes (ed Castegnaro et al.) International Agency for Research on Cancer (IARC) Publication No. 37.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}