![]()
![]()
![]()
Lilia Masson
La determinación de humedad es un paso obligado en el análisis de alimentos (1,2). Es la base de referencia que permite: comparar valores; convertir a valores de humedad tipo; expresar en base seca y expresar en base tal como se recibió.
Por estas razones debe seleccionarse cuidadosamente el método a aplicar para la determinación de humedad en un alimento, ya que un mismo método no sirve para todos los alimentos.
En general, los más usados aplican un cierto grado de calor. El alimento sufre cambios que pueden afectar el valor obtenido como humedad. Se pierden compuestos volátiles junto con el agua, como alcohol, aceites esenciales y materia grasa.
En el Cuadro N° 1 se encuentra un resumen de los métodos más usados para la determinación de humedad en alimentos, señalando brevemente sus ventajas, limitaciones y aplicaciones.
Los métodos más usados (3) son:
Requiere correcciones por el calor producido por solubilización de los óxidos de azufre y nitrógeno.
La calibración se realiza generalmente con ácido benzoico como patrón termoquímico.
Los valores obtenidos son los valores brutos de combustión y que deben diferenciarse de los valores de energía metabolizable, que habitualmente se emplean en nutrición.
La energía metabolizable se calcula aplicando los factores para: proteína, materia grasa, cabohidratos disponibles, ácidos orgánicos y alcohol.
En particular para fibra dietética, alcoholes azúcares, oligosacáridos, no hay aún acuerdo para estos componentes.
La expresión kilocalorías se sigue usando, a pesar que se recomienda el sistema internacional de kilojoule. El factor de conversión es 1 kcal = 4,184 kJ
Al expresar el valor de energía de un alimento deberá evitarse de usar más de tres cifras significativas. El método empleado cualquiera que sea debe ser descrito claramente.
Las siguientes consideraciones se deben tener en cuenta:
Cuadro 1
Métodos mas usados para la determinación de humedad
Energía bruta
Es la energía liberada como calor cuando el alimento se quema en la bomba calorimétrica. Se puede obtener por cálculo en base a la composición química del alimento utilizando los siguientes factores de conversión:
| - | Proteína: 5,4 kcal o 23 kJ/g |
| - | Carbohidrato: 4,1 kcal o 17 kJ/g |
| - | Grasa: 9,3 kcal o 39 kJ/g |
Energía disponible
Es la energía de los carbohidratos, grasas y proteínas menos la cantidad presente en las heces. A efectos de cálculo se utilizan los siguientes factores:
| - | Prote�nas: 5,4 -1,25 = 4,15. Se aproxima a 4,0 kcal o 17 kJ/g |
| - | Carbohidrato: 4,0 kcal o 17 kJ/g |
| - | Grasa: 9,0 kcal o 38 kJ/g |
Cuadro 2
Valores
de energía para algunos componentes de los alimentos
|
Componente |
kcal/g |
kJ/g |
|
Proteína |
4 |
17 |
|
Materia grasa |
9 |
37 |
|
Monosacárido |
3,75 |
16 |
|
Disacárido |
3,94 |
16 |
|
Almidón y glicógeno |
4,13 |
17 |
|
Alcohol etílico |
7 |
29 |
|
Glicerol |
4,31 |
18 |
|
Acido acético |
3,49 |
15 |
|
Acido cítrico |
2,47 |
10 |
|
Acido láctico |
3,62 |
15 |
|
Acido málico |
2,39 |
10 |
|
Acido quínico |
2,39 |
10 |
Problema de los carbohidratos
Otras tablas miden los carbohidratos disponibles, no calculan por diferencia. Calculados como monosacáridos usan 3,75 kcal o 16 kJ/g.
Fibra dietética
Se reduce la energía aparente disponible de grasa y proteína en 2-3% con ingestas moderadas y en un 4-6 % con ingestas altas
Se pueden aplicar varios métodos (1). A continuación se reseñará brevemente la base de ellos.
El contenido de etanol se puede medir en el destilado a partir de un volumen de la muestra exactamente medido.
Los azúcares reductores no son destilables. Interfieren el SO2 y el ácido acético a los niveles que se señalan a continuación:
Niveles de SO2
superiores a 200 mg/1
Acidez volátil que
exceda 0,10%
Por esta razón las muestras deben neutralizarse antes de la destilación. La determinación se efectúa por medio de un hidrómetro calibrado y se registra la
temperatura. El porcentaje de etanol en volumen se obtiene de tablas específicas.También se puede determinar el porcentaje de etanol en volumen con hidrómetros que miden el peso específico y consultar posteriormente las tablas pertinentes.
Los hidrómetros para etanol deben calibrarse con soluciones de etanol exactamente preparadas. Este método requiere bastante experiencia y cuidados extremos en todo el proceso de neutralización, destilación, medición y control de temperatura.
Requiere de un equipo especial. Mide el peso específico de la muestra, por el cambio de frecuencia de oscilación en un tubo en U comparado con dos estándares.
El peso específico se convierte a porcentaje de etanol a 15,56°C. Se aplica a destilados entre 25-79° alcohólicos.
El control de la temperatura y presión son factores importantes a considerar en la determinación.
Ocupa el refractómetro de inmersión. Debe tenerse un muy buen control de la temperatura a 15,56°C. A temperaturas
diferentes se aplican factores de corrección.Se utiliza el método del picnómetro. Se debe tener la precaución de un buen control de peso del picnómetro y bien calibrado, y un muy buen control de temperatura
El etanol obtenido por destilación se oxida cuantitativamente a ácido acético por un exceso de dicromato de potasio estandarizado.
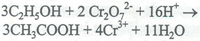
El exceso de K2Cr207 se determina con solución de sulfato ferroso y amonio estandarizado. Los tiempos y temperatura de incubación para oxidar el etanol a ácido acético son críticos.
Debe tenerse especial precaución con el material de vidrio, exactitud de las mediciones, calibración de pipetas, etc. Debe realizarse un blanco.
El etanol presente en vinos, cervezas, jugos se puede separar de otros componentes por GLC. Para mejorar
los aspectos cuantitativos se usa 2-propanol como estándar interno.La relación de área de ambos picos se compara con la de una solución patrón de etanol y 2-propanol.
Se pueden emplear columnas empacadas de acero inoxidable o vidrio de 2 m rellenas con Porapack QS, Carbowax 0,2 % 1500 sobre Carbopack C 80 -100 mesh, Chromosorb 103.
Se recomienda determinar el porcentaje de etanol en volumen por un método oficial para efectos de control.
El etanol se puede oxidar a acetaldehido por el NAD en presencia de la enzima alcohol dehidrogenasa (ADH) para producir NADH. Es una reacción esteoquiométrica en condiciones experimentales adecuadas. El NADH producido se determina espectrofotométricamente a 334, 340 o 360 nm.
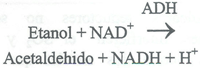
Actualmente se dispone de mezclas preparadas. Debe tenerse precaución en la medición cuantitativa de volúmenes muy pequeños de reactivo y muestra.
La reacción se desplaza a la derecha en medio alcalino, atrapando el acetaldehido formado y luego
oxidándolo a ácido acético en presencia de aldehido dehidrogenasa.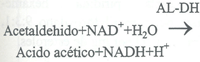
Dependiendo de la longitud de onda que se elija, se debe tener la precaución de que la concentración de etanol sea de 0,01 a 0,12g/l (365 nm) o de 0,005 a 0,06g/l(340o334nm).
En general en alimentos líquidos se recomienda clarificar y filtrar.
El método es muy sensible, debe tenerse especial cuidado con el agua empleada que debe estar libre de etanol. Igualmente se recomienda tapar las cubetas en el momento de la lectura. El etanol es muy volátil. Debe trabajarse en atmósfera libre de etanol. Para controlar el método se recomienda emplear una solución patrón de etanol.
Desde el punto de vista de una base de datos de composición de alimentos, el análisis y determinación de los diferentes componentes lipídicos de un alimento encierra una serie de desafíos y problemas.
Entre las determinaciones más frecuentes se pueden señalar:
| - | materia grasa total |
| - | materia insaponificable |
| - | esteroles |
| - | �cidos grasos |
| - | pigmentos carotenoides |
| - |
vitaminas liposolubles A, D, E, K |
Para cada uno de estos temas se dispone de diversas metodologías que se expondrán brevemente a continuación. Los dos últimos serán tratados en otra sección.
En el Cuadro 3 aparece un resumen de los métodos más empleados en la extracción de materia grasa total. Se ha señalado brevemente sus limitaciones, ventajas y aplicaciones.
Es un dato que normalmente se determina cuando se van a cuantificar esteroles u otros componentes que se encuentran en la materia insaponificable.
Métodos
| - | saponificación |
| - | avado |
| - | extracción con éter de petróleo, éter etílico |
| - | evaporación del solvente |
| - | pesada del residuo |
Limitaciones o inconvenientes
Debe asegurarse una completa saponificación, para lo cual la preparación del KOH alcohólico es importante.
Debe realizarse una cuidadosa extracción con los solventes.
Evitar los problemas de separación de fases, formación de emulsiones.
Realizar un lavado exhaustivo para retirar jabones.
Proceder a una cuidadosa evaporación del solvente y control del residuo final por pesada, que corresponde a la materia insaponificable.
a) Métodode la Comunidad Europea (6)
El método se basa en:
Saponificación de la materia grasa con KOH alcohólico, a la cual se adiciona alfa colestanol como patrón interno.
Extracción de la materia insaponificable con éter etílico.
Separación de la fracción de esteroles del insaponificable extraído mediante cromatografía en placa de sílica gel básica.
Los esteroles recuperados se derivatizan a trimetilsililéteres (TMSE) con mezcla piridina: hexametildisilazano: trimetilclorosilano 9:3:1, a razón de 50 TI por mg de esteroles.
Análisis por columna capilar GLC de sílica fundida o vidrio 20-30 m de largo de 0,25 a 0,32 mm diámetro interno, recubierta con fase líquida Se52, Se54 o equivalente, grosor de película entre 0,1 y 0,3 Tm y temperatura de la columna 260°C ± 5o.
Para la identificación de los diferentes picos se usan los tiempos de retención y la comparación con mezclas de TMSE de los esteroles analizados en las mismas condiciones.
El tiempo de retención del beta sitosterol se ajusta a 20±5 minutos.
Para la determinación cuantitativa se calculan las áreas de los picos del colestanol y de los esteroles dadas por el integrador y se aplica la fórmula para expresar en mg/kg de materia grasa.
b) Método general
La obtención de la materia insaponificable se puede realizar por método oficial AOAC, AOCS, Comunidad Europea (1,6).
Cuadro 3
Métodos para determinar
materia grasa total
| Método | Limitaciones | Ventajas y aplicaciones |
|
Extracción con un solvente |
||
| Continuo (BUTT) | Solventes inflamables Debe trabajarse sobre muestra seca o de muy baja humedad, menos de 8%. | Semillas oleaginosas, cubos de caldo saborizantes. Algunas sopas deshidratadas. |
| Discontinuo (SOXHLET) Eter etílico, éter de petroleo, alcoholes |
Extracción incompleta, usa alta temperatura, no puede usarse para estudio de ácidos grasos. No se puede aplicar a alimentos sometidos a algún tratamiento térmico ni a leche y derivados lácteos. | Algunos tipos de derivados cárneos, frutas y verduras, algunos productos azucarados. |
| Automático (FOSS-LET FAT ANALYZER) solvente Tetracloroetileno | Derivados cárneos | |
|
Extracción con mezcla de solventes |
||
| Hidrólisis acida previa a la extracción con solventes: Eter etílico, éter de petróleo | No se recomienda ocupar el extracto lipídico en el estudio de ácidos grasos aunque algunos autores lo emplean. No recomendable en alimentos ricos en azúcares, productos lácteos. | M�todo r�pido, de amplia aplicaci�n en diversos tipos de alimentos. Trabaja sobre muestra fresca. Debe aplicarse a todo alimento sometido a algun tipo de procesamiento o tratamiento t�rmico. |
| Hidrólisis alcalina previa a la extracción con solventes: Eter etílico, éter de petróleo. | Equipo especial con tubos y centrifuga adecuada. Alternativa: usar embudos de decantación. | Método de elección para leche y productos lácteos. |
| Método de Folch, Bligh y Dyer Cloroformo-Metanol | Cierto costo en solventes. Re-extracción para purificación, debe conocerse el % de humedad Debe conocerse el % de humedad del del alimento para ajustarla al 80%. | Extracción en frio, aplicable prácticamente a todo alimento. El extracto se puede usar para determinación de ácidos grasos,esteroles, etc. Son métodos rápidos. |
|
Métodos volumétricos |
||
| Carbonización selectiva con H2SO4 concentrado | Equipo especial | Rápido. Se usó mucho para leche y derivados lácteos. |
|
Otros métodos |
||
| Dispersión de la 1uz. Milkotester. | Equipo especial | Rápido. Leche cruda |
| Near Infrared Reflectance (NIR) | Equipo especial de alto costo. Requiere calibración |
Cereales. Leche. |
| NMR | Equipo especial de alto costo. Requiere calibración | Semillas oleaginosas. Método muy rápido. |
La derivatización puede realizarse para preparar acetatos, trimetilsililéteres, o butiratos.
El método de la Comunidad Europea purifica el residuo insaponificable por TLC antes de la derivatización. También se describen alternativas sin derivatizar.
El método es por cromatografía gas-líquida (GLC) en columnas de vidrio con fase OV 17, Se30, de 2 m, también se emplean columnas de sílica fundida de 12; 20; 30 m Se52, Se54 o equivalente.
El estándar interno puede ser 5 alfa colestano, o 5 alfa colestanol.
c) Método enzimático
Otro método para determinar colesterol es el método enzimático (7). El colesterol se oxida con colesterol oxidasa en presencia de agua oxigenada, catalasa y metanol, formándose finalmente una lutidina cuyo color se mide al espectrofotómetro a 405 nm. Las reacciones involucradas se señalan a continuación:

� formaldehído + acetilacetona + amonio
� Lutidina (el color se mide al espectrofotómetro a 405 nm).
El análisis de los ácidos grasos de cualquier alimento requiere de las siguientes etapas:
El primer punto ya fue tratado en métodos de extracción.
Se abordará a continuación los principales métodos de derivatización:
Al preparar los ésteres metílicos hay que considerar los siguientes aspectos que pueden afectar el resultado:
Los métodos de derivatización se pueden dividir en cuatro categorías (8):
| - | catálisis básica |
| - | catálisis ácida, básica |
| - | diazometano |
| - | otros métodos de esterificación |
a) Catálisis básica
Metóxido de sodio en metanol anhidro
Ventajas
Rápido, puede efectuarse a temperatura ambiente. Convierte los triglicerídos o acilgliceroles directamente a ésteres metílicos (transesterificación).
No produce isomerización de dobles enlaces. No libera el aldehido del plasmalógeno. No transesterifica ni esfingolípidos ni colesterol.
Desventajas
No convierte los ácidos grasos libres a ésteres metílicos. Por lo tanto no sirve para muestras de materias grasas con elevada acidez.
Debe trabajarse en medio anhidro. La presencia de agua produce saponificación lo cual resulta en
pérdida de ácidos grasos. El uso prolongado puede alterar la composición en ácidos grasos.Alta concentración de álcali y alta temperatura puede llevar a la formación de ácidos grasos conjugados.
HC1 en metanol 5%
P/V
H2SO4 en metanol 1 a 2% V/V
Cloruro de acetilo en metanol
KOH en metanol y BF3 en metanol
Ventajas
Todos estos métodos convierten los acilgliceroles o triglicéridos y ácidos grasos libres en ésteres metílicos. El más popular tal vez es el BF3 20% en metanol por usar el primer procedimiento aceptado por AOCS.
El método de la AOAC saponifica con NAOH en metanol y luego esterifica con BF3 en metanol. Es un método rápido.
Desventajas
Forma compuestos ajenos o interferentes.
No es útil para aceites de semilla que contienen ácidos grasos inusuales como epoxi, ciclopropeno, insaturación conjugada, etc.
Reacciona con plasmalógenosformando aldehido.
Reacciona con colesterol formando colesterolmetiléster y colestadieno.
El reactivo es caro e inestable. Debe refrigerarse. El empleo de BF3 antiguo o concentrado produce pérdidas de ácidos grasos poliinsaturados.
Cloruro de acetilo en metanol
Ventajas
Presenta buenos resultados con leche y aceites vegetales sin extracción previa de los lípidos.
Comparable al BF3-metanol para transesterifícar los colesteril ésteres, no produce compuestos extraños.
Desventaja
No esterifica los ácidos grasos libres
c) Diazometano
Ventajas
Es un método rápido casi instantáneo a temperatura ambiente.
Esterifica los ácidos grasos libres en presencia de metanol.
El exceso de reactivo se elimina por evaporación bajo N2.
Desventaja
Reactivo muy tóxico. Se puede usar sólo si es estrictamente necesario. Es explosivo. Forma compuestos extraños reaccionando con dobles enlaces o grupos carbonilos.
Se han buscado nuevos reactivos con el objeto de obtener mas rápidamente los ésteres metílicos de los ácidos grasos.
1. Utilización, de sales cuaternarias de amonia
Se han usado sales cuaternarias de amonio fuertemente básicas como:
| - | hidróxido de m-trifluorometilfeniltrimetilamonio (TFMPTAH) |
| - |
hidróxido de trimetilfenilamonio (TMPAH) |
| - |
hidróxido de trimetilamonio (TMPAH) |
Ventajas
Se produce la transesterificación de los acilgliceroles o triglicéridos a. ésteres metílicos de ácidos grasos por pirólisis en la columna GLC.
La transesterificación se efectúa en una etapa, no se requiere etapa de
extracción. De especial importancia en ácidos grasos de cadenas cortas.Se podría determinar separadamente la composición en ácidos grasos de los triglicéridos, de los ácidos grasos libres y del total.
Desventajas
La muestra debe ser neutralizada antes del análisis por GLC. La alta alcalinidad y alta temperatura de la columna para la metilación pirolítica produce isomerización de dobles enlaces dando ácidos grasos conjugados. Algunos productos de ruptura de estos reactivos pueden interferir con el análisis de ácidos grasos de cadena mediana.
2. Transesterificación directa
Cloruro de acetilo en metanol
Ventajas
La extracción y aislación del lípido se efectúa en una sola etapa.
La muestra se coloca en un solvente, se agrega cloruro de acetilo en metanol y se obtiene los ésteres metílicos de los ácidos grasos. Se han investigado diferentes solventes.
Se ha aplicado a tejidos, plasma, alimentos, leche, grasa de leche, etc. Se ha mostrado más eficiente comparado
con el método tradicional de extracción de Folch.3. Metanol caliente en HCL
Ventaja
Se aplicó a hojas y los ésteres metílicos de los ácidos grasos se extrajeron con solvente.
Desventaja
Se obtuvieron valores más bajos que los obtenidos por métodos convencionales.
Guanidina y sus derivados alquilados en metanol
Ventajas
Se produce una completa metanólisis de los aceites y se convierte los acilgliceroles o triglicéridos y ácidos grasos libres en ésteres metílicos.
Los ácidos grasos forman la sal guanidinacarboxilato que en presencia de exceso de metanol forma los ésteres metílicos.
Este método se ha comparado satisfactoriamente con el método Ce 2-66 con 20% BF3 en metanol de la AOCS.El reactivo no es caro, no produce isomerización de dobles enlaces. Es un
método rápido, bastan 2 minutos en baño de agua hirviendo.Desventaja
El exceso de reactivo debe ser extraído antes que la muestra se inyecte en la columna GLC.
4. Metilación con metanol catálisis básica y ácida
La metilación se efectúa con metilato de sodio, calentando a reflujo 10 minuos. Se enfría y luego se agrega la solución metanólica de H2SO4 hasta pH ácido, viraje de la fenolftaleína, luego se calienta a reflujo por 20 minutos, se enfría. Se agrega el hexano para la extracción de los ésteres metílicos.
5. Preparación de ésteres butílicos
Se prefiere para el análisis de ácidos grasos de cadena corta presentes en grasa de leche.
Se usa BF3 al 12,5 % en butanol
Ventaja
Da mejores recuperaciones de estos ácidos grasos de cadena corta.
Preparación de ésteres metílicos
La Comunidad Europea (9) señala los siguientes métodos para la preparación de los ésteres metílicos.
Recomendación: no se tomará en cuenta la materia insaponificable cuando no supere el 3%. En caso contrario, los ésteres metílicos se prepararán a partir de los ácidos grasos.
Cromatografía gas-líquido de los
ésteres metílicos. Método de la Comunidad Europea(9)Señala las siguientes alternativas:
Otras consideraciones en relación a la cromatografía gas-líquido de ésteres metílicos de ácidos grasos
El Dr. Robert Ackman ha propuesto que las fases de tipo Carbowax 20M sean utilizadas como patrones de referencia para columnas capilares de sílica fundida en ensayos interlaboratorios. También ha trabajado bastante con Silar CP, una fase que es ligeramente más polar que Carbowax 20M. FFAP, Supelcowax-10 y SP 1000 son muy similares al Carbowax 20M en columnas de sílica fundida.
Fases estacionaria con enlace cruzado y unidas (bonded) se han mostrado satisfactorias en la resolución de ciertos pares críticos.
En el caso de columnas empacadas, tres columnas que contienen 15% EGSS-Y, EGSS-X y Silar 10C sobre soporte silanizado 100-120 mesh, lavado al ácido abren un amplio rango de polaridad para el análisis de la mayoría de los ácidos grasos poliinsaturados.
Gas portador
El hidrógeno tiene ventajas como gas portador para las columnas capilares de tubo abierto en términos de eficiencia. Problema: detectar pérdidas o escapes permanentemente por seguridad. El helio es más seguro pero más costoso en diversos países.
Los gases deben pasarse a través de trampas de oxígeno y humedad antes de la columnas para dar mayor estabilidad a la línea base cuando se trabaja a altas sensibilidades y para prolongar la vida de la columna. Para columnas empacadas, nitrógeno con menos de 5 ppm de oxígeno es adecuado. El argón es más caro que el nitrógeno y contiene menos oxígeno.
El horno
Deberá tener un dispositivo altamente sensible para controlar la temperatura,
de modo que sea uniforme en todas las regiones.Uso de patrones para identificación
Existen en el mercado mezclas patrones conteniendo cantidades conocidas de ésteres metílicos de ácidos grasos saturados, monoenoicos y polienoicos. Sirven para efectos de identificación y cuantificación.
Para identificación se usan los tiempos de retención corregidos o relativos de los patrones en relación a los componentes de la muestra, corridos en la misma columna y bajo idénticas condiciones. La comparación se debe hacer en dos columnas con fases líquidas de polaridad diferente. El largo de cadena equivalente es otra posibilidad de identificación.
Como algunos lípidos de origen animal y marino contienen mezclas complejas de ácidos grasos, se recomienda usar algunas materias grasas de composición conocida como: aceite de hígado de bacalao, materia grasa de testículos de bovino y porcino, lípidos de hígado de rata, etc.
Valores para ácidos grasos
Los valores para ácidos grasos individuales, generalmente se expresan como porcentaje de ácidos grasos en relación al total, ya que esta es la forma más común de presentación analítica.
A nivel de base de datos se necesita la información por 100 g de alimento. Por lo tanto, se han publicado factores de conversión derivados de la proporción de los ácidos grasos presentes en el lípido total para convertir porcentajes de ácidos grasos a ácidos grasos por 100 g de alimento (3).
Conclusiones generales
![]()
![]()
![]()